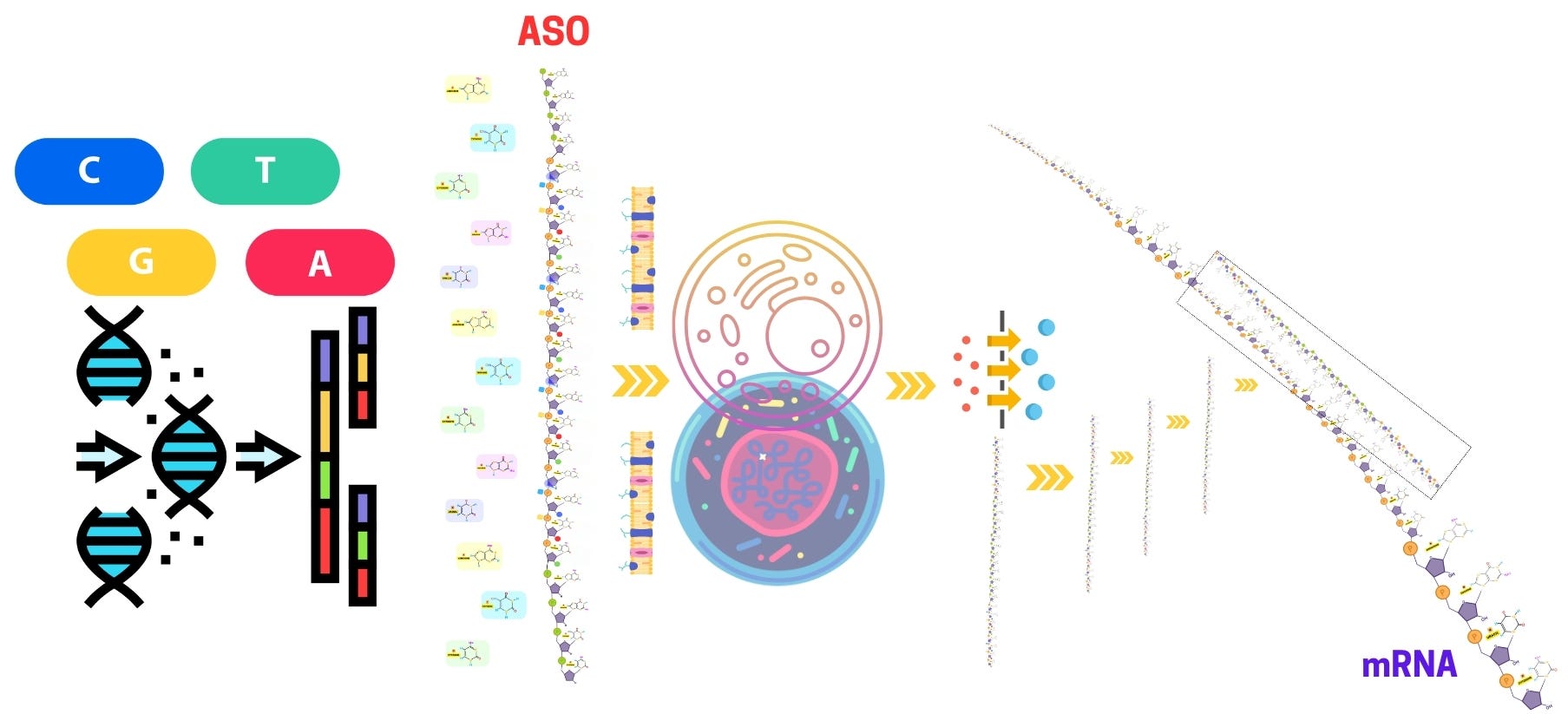
Antisense oligonucleotides (ASOs) have emerged as a groundbreaking approach for gene silencing and modulating gene expression, offering promising therapeutic potential for genetic disorders, neurodegenerative diseases, and various cancers. The development of antisense oligonucleotide therapy has fueled extensive research into antisense oligo modifications, delivery mechanisms, and strategies to enhance stability and specificity. Key advancements include phosphorothioate oligonucleotides, PMO oligos, and gapmer antisense oligonucleotides, which improve clinical efficacy by increasing resistance to nuclease degradation.
In parallel, the rise of antisense RNAi has broadened the landscape of gene-silencing approaches, working alongside traditional ASO methods to expand therapeutic applications. Emerging technologies such as locked nucleic acid (LNA) antisense oligonucleotides and morpholino antisense oligos further optimize target engagement while minimizing off-target effects. Additionally, antisense oligonucleotide synthesis continues to evolve, incorporating chemical modifications like 2'-O-methyl phosphorothioate ASOs to enhance RNA targeting efficiency. As research progresses, these innovations pave the way for more effective ASO-based treatments, addressing unmet medical needs in diseases with limited treatment options.
The chemical modification of antisense oligonucleotides
The chemical modification of antisense oligonucleotides (ASOs) is a critical aspect of their design, enhancing their biochemical properties and therapeutic potential. ASOs, designed to hybridize with specific RNA targets to modulate gene expression, are inherently unstable in biological environments and prone to rapid degradation by nucleases. Moreover, unmodified ASOs can suffer from poor pharmacokinetics, off-target effects, and immune activation, which significantly limits their therapeutic window. To overcome these challenges, a wide array of chemical modifications has been developed to improve the stability, binding affinity, target specificity, and pharmacokinetic profiles of ASOs.
Reverse Phosphoramidites, phosphoramidite group is attached to the 5' hydroxyl end of the nucleoside, formation of non-standard linkages, such as 3'-3' and 5'-5' linkages
Backbone modifications (e.g., phosphorothioate) and their impact on stability.
Sugar modifications (e.g., 2'-OMe, LNA) for increased binding affinity.
End modifications (e.g., capping) to protect ASOs from exonuclease degradation.
Chemical Conjugates for Antisense Oligonucleotides (ASOs) Their Targets & Efficacy
Chemistry in ASO Ligand Conjugation (NHS vs Click)
The landscape of chemical modifications for ASOs is vast and continually evolving. These modifications are crucial for enhancing the therapeutic potential of ASOs by:
Improving Stability: Against nucleases in biological environments.
Enhancing Affinity: For target RNA or DNA sequences.
Reducing Immunogenicity: Minimizing immune system activation.
Facilitating Delivery: Improving cellular uptake and tissue targeting.
Modulating Pharmacokinetics: Affecting distribution, metabolism, and excretion.
Key modification strategies target the ASO backbone, sugar moieties, and terminal ends, each contributing to different aspects of ASO performance. Backbone modifications, such as the introduction of phosphorothioate (PS) linkages, confer enhanced resistance to both exonucleases and endonucleases, extending the ASO's half-life in biological fluids. Phosphorothioates also improve the ASO’s interaction with plasma proteins, particularly albumin, thereby increasing circulation time and reducing renal clearance. However, these modifications can introduce stereochemical complexity, as sulfur atoms create diastereomers with varying biological effects, necessitating careful design to mitigate potential drawbacks.
Sugar modifications, particularly at the 2' position of the ribose, significantly enhance the binding affinity and stability of ASO-RNA duplexes. Modifications like 2'-O-methyl (2'-OMe) and 2'-O-methoxyethyl (2'-MOE) not only stabilize the ASO-RNA interaction by increasing the thermal stability of the duplex but also protect the ASO from enzymatic degradation, particularly RNase H-mediated cleavage. Locked Nucleic Acids (LNA), another advanced sugar modification, dramatically increase the binding affinity due to their rigid structure, allowing for shorter and more potent ASOs with enhanced specificity. LNAs are particularly advantageous in therapeutic settings where high-affinity binding and nuclease resistance are required; however, their use must be carefully optimized to avoid potential cytotoxicity associated with over-modification.
End modifications, including 3' and 5' capping strategies, are equally important for protecting ASOs from degradation by exonucleases. Common strategies, such as capping with inverted thymidine or phosphorothioate-modified nucleotides, provide protection while maintaining hybridization efficiency with the target RNA. Additionally, conjugation of ASOs with polyethylene glycol (PEG) or cholesterol at the terminal ends enhances solubility, reduces immune activation, and improves tissue distribution, further extending the ASO’s half-life in vivo and enhancing delivery to target tissues. These end modifications are particularly important in optimizing ASOs for systemic administration, where prolonged circulation and improved biodistribution are required.
This article delves into the technical details of these chemical modifications, outlining the impact of each modification on the pharmacokinetics, stability, and efficacy of ASOs. It also highlights emerging strategies, such as novel backbone modifications (e.g., phosphorodiamidate morpholino oligomers) and sugar modifications (e.g., 2'-fluoro and tricyclic DNA), which further refine ASO design by offering enhanced specificity, stability, and target engagement. These evolving chemical strategies form the basis for next-generation ASO therapeutics, allowing researchers to fine-tune ASOs for a variety of therapeutic applications, from exon skipping and translation inhibition to RNA degradation and splicing modulation.
In conclusion, chemical modification is a crucial component of ASO development, enabling the creation of oligonucleotides that are biochemically robust, highly specific, and therapeutically effective. By strategically altering the backbone, sugar moieties, and terminal ends of ASOs, researchers can optimize their pharmacological properties to meet the demands of clinical applications, pushing the boundaries of gene modulation therapies.
Chemical Modification of ASOs
Chemical modification is crucial for enhancing ASO stability, binding affinity, and resistance to enzymatic degradation. Common backbone modifications, such as phosphorothioate (PS) groups, are introduced to increase resistance to nucleases and improve pharmacokinetic properties. Sugar modifications, like 2'-O-Methyl (2'-OMe) or Locked Nucleic Acids (LNA), are employed to boost binding affinity and further protect the ASO from degradation. End modifications, such as capping, help prevent exonuclease-mediated degradation, ensuring that the ASO remains intact long enough to reach its target.
Chemical modification is a pivotal step in the development of antisense oligonucleotides (ASOs) that enhances their stability, specificity, binding affinity, and overall efficacy. Natural oligonucleotides are prone to rapid degradation by nucleases, have limited bioavailability, and may induce off-target effects or immune responses. Chemical modifications address these challenges, tailoring ASOs to improve their pharmacokinetics, tissue distribution, and biological performance. In this section, we will explore the technical aspects of backbone, sugar, and end modifications, and how they contribute to the design of therapeutic ASOs.
Reverse Phosphoramidites
In oligonucleotide synthesis, the phosphoramidite approach is the gold standard, enabling the assembly of DNA and RNA sequences by adding nucleotides sequentially in the 3'→5' direction. However, specialized applications require modifications to this conventional approach. One such modification involves the use of Reverse Phosphoramidites, where the phosphoramidite group is attached to the 5' hydroxyl end of the nucleoside, facilitating synthesis in the 5'→3' direction or enabling the formation of non-standard linkages, such as 3'-3' and 5'-5' linkages.
Conventional Phosphoramidite (3' → 5'):
5' Phosphate — Nucleotide Base — OH 3' | Phosphoramidite
Phosphoramidite is attached to the 3' hydroxyl of the nucleoside.
Reverse Phosphoramidite (5' → 3'):
5' OH — Nucleotide Base — Phosphoramidite — O
Phosphoramidite is attached to the 5' hydroxyl of the nucleoside.
The 5' hydroxyl esterifies with the phosphoramidite group, while the 3' end remains available for nucleotide coupling.
The Chemistry Behind Reverse Phosphoramidites
Traditional phosphoramidites have the phosphoramidite group attached to the 3' hydroxyl of the nucleoside. This allows the sequential addition of nucleotides to occur from 3' to 5', the natural direction of DNA/RNA synthesis in vivo. In contrast, reverse phosphoramidites are designed with the phosphoramidite group on the 5' hydroxyl. This alteration opens up two main possibilities:
Reverse synthesis in the 5'→3' direction: While the conventional synthesis proceeds in the 3'→5' direction, reverse phosphoramidites enable the chain to elongate in the 5'→3' direction. This is critical in scenarios where the oligonucleotide needs to be synthesized with an inverted order or structure.
Inverted Linkages (3'-3' and 5'-5'): Another feature of reverse phosphoramidites is their ability to create non-standard linkages, such as 3'-3' or 5'-5' bonds. These inverted linkages are useful for creating specific structures such as cyclic oligonucleotides, branched DNA/RNA structures, or for stabilizing certain segments of the sequence.
Reverse phosphoramidites represent a powerful tool in the synthesis of oligonucleotides, allowing for the 5'→3' synthesis direction and enabling the formation of inverted linkages such as 3'-3' or 5'-5'. These capabilities are essential for applications in antisense therapy, nanotechnology, and molecular diagnostics, providing researchers with greater flexibility to design oligonucleotides with unique structures and functionalities. However, as with any advanced chemical synthesis, challenges related to efficiency, stability, and purification need to be carefully managed to ensure optimal results.
Including reverse phosphoramidites in an antisense oligonucleotide (ASO) sequence can provide specific advantages and structural modifications. Here are the key applications and uses:
Inverted Linkages for Increased Stability
3'-3' or 5'-5' Linkages: By incorporating reverse phosphoramidites, you can create inverted 3'-3' or 5'-5' linkages within the ASO sequence. These non-canonical linkages can increase the overall stability of the ASO, making it more resistant to nuclease degradation in biological environments. This is especially valuable in therapeutic applications where maintaining ASO integrity is crucial.
Enhancing Binding Specificity and Affinity
Improved Binding: Introducing reverse phosphoramidites can optimize the ASO’s hybridization properties by altering the orientation of nucleotides. This could help in fine-tuning the duplex stability with the target RNA, improving binding specificity and affinity, and ensuring more precise targeting of the mRNA of interest.
Blocking Exonuclease Activity
Protecting End Groups: Reverse phosphoramidites can be added at the 5' end of the ASO to block exonuclease-mediated degradation. This terminal modification can serve as a protective mechanism to extend the ASO's half-life in biological systems, thus increasing its therapeutic efficacy.
Cyclic Oligonucleotide Structures
Cyclic ASO Design: Reverse phosphoramidites enable the formation of cyclic ASO structures by creating head-to-tail linkages (5'–3' or 3'–5' cyclic configurations). Cyclic oligonucleotides offer unique benefits, such as enhanced binding specificity, increased resistance to degradation, and improved cellular uptake. These structures are useful in drug delivery and molecular probes.
Branching in ASO Design
Multivalent or Branched ASOs: Reverse phosphoramidites can introduce branching points, creating multivalent ASOs. These branched structures can be designed to target multiple regions of the same RNA or different RNA molecules, potentially increasing therapeutic potency by engaging multiple binding sites.
Tunable Degradation Profile
Controlled Degradation: By carefully placing reverse phosphoramidites in specific regions of the ASO sequence, you can design ASOs with a tunable degradation profile. This could allow for controlled release of the ASO’s active segments, ensuring that it remains effective over a longer period of time.
Improved Pharmacokinetics and Biodistribution
Prolonged Circulation: ASOs modified with reverse phosphoramidites at key positions can improve pharmacokinetics by reducing rapid clearance and improving biodistribution. This makes reverse-modified ASOs ideal candidates for therapeutic use, where longer circulation times in the bloodstream are desired.
Increased Sequence Diversity
Expanding ASO Design Options: The inclusion of reverse phosphoramidites increases the structural diversity of ASOs, allowing for innovative designs that can address different therapeutic challenges. For example, reverse ASOs can potentially target unconventional RNA structures or disease-causing mutations that are difficult to target with traditional ASO designs.
Modulating Cellular Uptake and Distribution
Altering Charge Distribution: The incorporation of reverse phosphoramidites may change the charge distribution and hydrophobicity of the ASO, which could influence its ability to penetrate cellular membranes and its distribution within tissues. This can be critical in targeting specific tissues or cell types.
Dual-Function ASOs
Functionalization: Reverse phosphoramidites can facilitate the attachment of functional groups or ligands, such as targeting molecules, fluorophores, or other chemical moieties. This enables the creation of ASOs that not only bind target RNA but also carry additional functionality, such as targeted delivery or imaging capabilities.
Backbone Modifications
The backbone of an oligonucleotide consists of phosphate linkages between the sugar molecules of adjacent nucleotides. In unmodified ASOs, the natural phosphodiester (PO) backbone is highly susceptible to degradation by nucleases, limiting the therapeutic window. Modifying the backbone structure improves resistance to enzymatic degradation and modulates pharmacokinetics and cellular uptake.
Phosphorothioate (PS) Linkages
Phosphorothioate (PS) modification is one of the most commonly employed backbone modifications in ASO design. In PS-modified ASOs, one of the non-bridging oxygen atoms in the phosphate group is replaced by sulfur, creating a phosphorothioate bond.
Nuclease Resistance: PS linkages significantly enhance the ASO’s resistance to exonucleases and endonucleases, prolonging the ASO’s half-life in biological fluids. The sulfur atom reduces the affinity of nucleases for the phosphate backbone, preventing cleavage.
Pharmacokinetics: PS-modified ASOs exhibit improved pharmacokinetic profiles due to their enhanced stability. The PS backbone also increases plasma protein binding, particularly to albumin, which extends the ASO’s circulation time and reduces renal clearance.
Challenges: While PS modifications improve stability, they can introduce some drawbacks. PS linkages slightly reduce binding affinity compared to unmodified phosphodiester linkages. Furthermore, the introduction of sulfur atoms introduces chirality at each modified phosphate bond, leading to the formation of R and S diastereomers. This heterogeneity can impact the ASO’s efficacy and toxicity.
Phosphoramidate and Phosphorodiamidate Modifications
Other backbone modifications include phosphoramidate and phosphorodiamidate linkages, which provide additional protection against nucleases.
Methylphosphonates: In methylphosphonate-modified ASOs, a methyl group replaces one of the non-bridging oxygens in the phosphate backbone. This modification enhances nuclease resistance while maintaining binding affinity. However, methylphosphonate-modified ASOs tend to have poor water solubility, limiting their use in some therapeutic applications.
Phosphorodiamidate Morpholino Oligomers (PMOs): PMOs are a type of ASO where the ribose sugar and phosphate backbone are replaced with morpholine rings and phosphorodiamidate linkages. PMOs are highly resistant to nucleases and have improved stability in biological fluids. They are commonly used in exon-skipping therapies, such as those for Duchenne muscular dystrophy (DMD).
Sugar Modifications
The sugar moiety in the backbone of an oligonucleotide is responsible for maintaining the structural integrity of the molecule and influencing binding affinity. Modifications to the sugar ring enhance the ASO's stability, affinity for the target RNA, and reduce immunogenicity. These modifications mainly involve the ribose sugar's 2' position, where hydroxyl groups are replaced with functional groups to improve various properties.
2'-O-Methyl (2'-OMe) and 2'-O-Methoxyethyl (2'-MOE) Modifications
2'-O-alkyl modifications replace the hydroxyl group at the 2' position of the ribose sugar with small alkyl groups, improving stability and binding affinity.
2'-O-Methyl (2'-OMe): The 2'-OMe modification is widely used in ASOs to increase the affinity between the ASO and its target RNA. The presence of the 2'-OMe group enhances the thermal stability of the ASO-RNA duplex, thereby increasing binding specificity. Additionally, 2'-OMe modifications improve nuclease resistance, particularly against RNase H degradation, extending the ASO’s half-life.
2'-O-Methoxyethyl (2'-MOE): The 2'-MOE modification is a more potent version of the 2'-OMe modification, providing even greater stability and affinity. 2'-MOE-modified ASOs exhibit higher melting temperatures (Tm), indicating stronger binding to the target RNA. This modification is often used in therapeutic ASOs because it balances high affinity with reduced toxicity and immunogenicity.
Locked Nucleic Acids (LNA)
Locked nucleic acids (LNAs) are a class of chemically modified nucleotides in which the ribose sugar is "locked" into a rigid conformation by forming a methylene bridge between the 2' oxygen and the 4' carbon of the sugar ring.
Increased Binding Affinity: LNA modifications dramatically enhance binding affinity to the target RNA by stabilizing the ASO-RNA duplex. The rigid conformation of the LNA structure increases the duplex's melting temperature (Tm) by up to 10°C per LNA residue incorporated into the ASO, making LNAs highly attractive for therapeutic applications.
Improved Specificity: The increased affinity of LNAs also improves the specificity of the ASO, reducing the risk of off-target effects. This heightened specificity allows the use of shorter ASOs without sacrificing efficacy, further minimizing potential off-target interactions.
Nuclease Resistance: LNAs offer superior resistance to exonuclease degradation, making them ideal for applications where long-term stability is required.
Challenges: Despite their advantages, LNA-modified ASOs can induce toxicity, particularly when multiple LNA residues are incorporated. Careful optimization of the number and position of LNA residues in the ASO is necessary to balance efficacy and safety.
2'-Fluoro (2'-F) and 2'-O-Alkyl Modifications
Other sugar modifications that enhance ASO performance include 2'-fluoro and 2'-O-alkyl modifications.
2'-Fluoro (2'-F): In 2'-F modifications, the hydroxyl group at the 2' position is replaced with a fluorine atom. This modification increases binding affinity and stability, similar to 2'-OMe and 2'-MOE modifications. 2'-F-modified ASOs have been shown to induce potent RNase H activity, making them particularly effective in gene silencing applications.
2'-O-Alkyl Modifications: In these modifications, larger alkyl groups, such as ethyl or isopropyl, are attached to the 2' position of the ribose. These bulky groups provide enhanced nuclease resistance and stability while maintaining binding affinity.
End Modifications
End modifications, particularly at the 3' and 5' ends of the ASO, are essential for preventing exonuclease degradation and improving overall stability. These modifications ensure that the ASO remains intact long enough to exert its therapeutic effect in vivo.
3'-End Capping
The 3' end of the ASO is particularly vulnerable to degradation by 3'-exonucleases. To prevent this, chemical groups are added to cap the 3' terminus.
Inverted Thymidine (InT): A commonly used cap is the addition of an inverted thymidine residue at the 3' end. This modification prevents exonuclease recognition and degradation without significantly affecting the ASO’s hybridization to its target RNA.
Phosphate or Phosphorothioate Capping: Adding a 3'-phosphate group or phosphorothioate modification also helps protect the ASO from exonuclease activity. These capping strategies improve the ASO’s stability in biological fluids while maintaining its functionality.
5'-End Modifications
The 5' end can also be capped to protect against 5'-exonucleases, although this is less common than 3' capping.
PEGylation: Adding polyethylene glycol (PEG) to the 5' end of the ASO can improve solubility, reduce immunogenicity, and increase circulation time in vivo. PEGylated ASOs exhibit enhanced pharmacokinetic properties due to their ability to evade immune detection and clearance by the kidneys.
Cholesterol Conjugation: Attaching a cholesterol group to the 5' end can enhance the ASO’s ability to cross cell membranes and improve intracellular delivery. Cholesterol conjugation is often used in combination with other delivery systems, such as lipid nanoparticles, to improve bioavailability.
Novel and Emerging Chemical Modifications
As the field of ASO therapeutics continues to evolve, new chemical modifications are being developed to further enhance the properties of ASOs. Some promising modifications include.
Peptide Nucleic Acids (PNAs)
Peptide nucleic acids (PNAs) are synthetic analogs of DNA or RNA in which the sugar-phosphate backbone is replaced by a peptide-like backbone composed of repeating N-(2-aminoethyl)-glycine units. This modification confers several advantages.
Improved Stability: PNAs are highly resistant to nucleases and proteases, making them extremely stable in biological systems.
High Affinity: PNAs form very stable duplexes with complementary RNA or DNA due to their neutral backbone, which reduces electrostatic repulsion during hybridization.
Challenges: Despite their stability and high affinity, PNAs have poor water solubility and cell permeability, necessitating the use of delivery vehicles such as liposomes or nanoparticles for therapeutic applications.
Tricyclic DNA (tcDNA)
Tricyclic DNA is a novel modification that introduces rigid, tricyclic structures into the ASO backbone, further enhancing binding affinity and specificity.
Increased Affinity: tcDNA-modified ASOs exhibit an even higher binding affinity than LNA-modified ASOs, making them promising candidates for therapeutic applications where strong binding is required.
Enhanced Specificity: The rigidity of the tricyclic structure improves the selectivity of the ASO for its target RNA, reducing off-target interactions.
Challenges: As with LNA, the incorporation of tcDNA residues must be carefully optimized to avoid potential toxicity.
ASO Ligand Conjugates
To enhance their therapeutic potential, ASOs are often chemically conjugated to various molecules that improve their pharmacokinetic properties, cellular uptake, and tissue-specific delivery and their advantages in terms of tissue delivery mechanisms, efficacy, and cell type specificity.
N-Acetylgalactosamine (GalNAc) Conjugation
Description: GalNAc moieties are conjugated to ASOs to target the asialoglycoprotein receptor (ASGPR) on hepatocytes.
Tissue Delivery Mechanism:
Liver Targeting: ASGPR is highly expressed on hepatocytes. GalNAc binds specifically to ASGPR, facilitating receptor-mediated endocytosis.
Efficient Internalization: The GalNAc-ASO complex is rapidly internalized into hepatocytes after binding.
Efficacy:
Enhanced Potency: Increases ASO accumulation in the liver, improving therapeutic outcomes.
Targeted Delivery: Enhances ASO concentration in disease-relevant tissues.
Therapeutic Synergy: Ligand may have independent therapeutic effects.
Cell Type Specificity:
Tumor Cells: Overexpress certain receptors like sigma or integrins.
Angiogenic Vasculature: Targeting new blood vessels in tumors.
Dendrimer Conjugation
Description: ASOs are attached to dendrimers—highly branched macromolecules.
Tissue Delivery Mechanism:
Multivalent Interaction: Multiple ASOs on one dendrimer increase binding to cell surfaces.
Enhanced Permeability: Dendrimers can facilitate passage through biological barriers.
Efficacy:
Increased Loading Capacity: Dendrimers can carry multiple ASO molecules.
Protection from Degradation: Dendrimer structure shields ASOs from nucleases.
Cell Type Specificity:
Tumor Targeting: Functionalization with targeting ligands can direct dendrimers to cancer cells.
Potential for CNS Delivery: Certain dendrimers can cross the BBB.
Oligosaccharide Conjugation
Description: Conjugation with specific oligosaccharides to target carbohydrate-binding receptors.
Tissue Delivery Mechanism:
Lectin Receptor Binding: Targets cells expressing specific lectins.
Facilitated Uptake: Oligosaccharides can enhance cellular internalization.
Efficacy:
Improved Targeting: Enhances delivery to cells of interest.
Potential Immunomodulation: Oligosaccharides can influence immune cell behavior.
Cell Type Specificity:
Liver and Immune Cells: Express various lectin receptors.
Applications in Immunotherapy: Potential for targeting dendritic cells.
Biotin-Streptavidin Conjugation
Description: ASOs are biotinylated and complexed with streptavidin-linked targeting agents.
Tissue Delivery Mechanism:
Versatile Targeting: Streptavidin can be linked to various molecules, allowing for customizable delivery.
Receptor-Mediated Uptake: Dependent on the targeting agent attached to streptavidin.
Efficacy:
High Affinity Interaction: Biotin-streptavidin binding is strong, ensuring stability of the conjugate.
Enhanced Delivery: Targeting agent directs ASO to specific cells.
Cell Type Specificity:
Customizable: Specificity depends on the chosen targeting molecule.
Research Tool: Commonly used in experimental settings for proof-of-concept studies.
Sialic Acid Conjugation
Description: ASOs conjugated with sialic acid to target Siglec receptors.
Tissue Delivery Mechanism:
Siglec Binding: Sialic acid binds to sialic acid-binding immunoglobulin-like lectins on immune cells.
Receptor-Mediated Uptake: Facilitates internalization into Siglec-expressing cells.
Efficacy:
Immune Cell Modulation: Direct delivery to immune cells can modulate immune responses.
Reduced Immunogenicity: Sialic acid may help in evading immune detection.
Cell Type Specificity:
B Cells and Macrophages: Express various Siglecs.
Applications in Autoimmune Diseases: Potential to regulate overactive immune cells.
Polymer Conjugation (Other than PEG)
Description: ASOs conjugated with polymers like polyamidoamine (PAMAM) dendrimers or poly(L-lysine).
Tissue Delivery Mechanism:
Enhanced Uptake: Polymers can facilitate cellular entry via endocytosis.
Controlled Release: Polymers can be designed to release ASOs under specific conditions.
Efficacy:
Protection from Degradation: Polymers shield ASOs from nucleases.
Increased Payload: Allows for the delivery of multiple ASOs or combination therapies.
Cell Type Specificity:
Tumor Targeting: Polymers can be modified with targeting ligands.
Potential for Gene Therapy: Useful in delivering genetic material to specific cells.
Chemical conjugation of ASOs enhances their therapeutic potential by improving delivery mechanisms, efficacy, and cell type specificity. By selecting appropriate conjugates, ASOs can be tailored to target specific tissues or cell types, reduce systemic side effects, and achieve better clinical outcomes. The diversity of conjugation strategies allows for customization based on the disease target, desired pharmacokinetic profile, and therapeutic goals.
Chemistry in ASO Ligand Conjugation
NHS ester chemistry and click chemistry are commonly used methods for adding conjugates to antisense oligonucleotides (ASOs), as well as other types of biomolecules. These conjugation methods are widely applied in bioconjugation processes for various purposes such as enhancing cellular uptake, improving stability, and enabling targeted delivery.
NHS Ester Chemistry in ASO Conjugation:
Primary use: NHS esters can be used to attach small molecules, peptides, or other targeting moieties to the primary amine groups present on ASOs (or on linkers attached to ASOs).
Application: This method is particularly suitable for adding functionalities like fluorophores for imaging, or conjugating cell-penetrating peptides (CPPs) or lipids to improve cellular uptake.
Limitations: Because NHS ester chemistry reacts with any available primary amine, it may lead to non-specific reactions if not well-controlled, especially in complex environments.
Click Chemistry in ASO Conjugation:
Primary use: Click chemistry, especially the copper-free variant (e.g., strain-promoted azide-alkyne cycloaddition, SPAAC), is highly favored for conjugating ASOs due to its specificity, high efficiency, and bioorthogonality.
Application: Click chemistry is used to attach ligands (such as antibodies, aptamers, or other targeting molecules) that direct ASOs to specific cell types or tissues. It is also used to conjugate ASOs with nanoparticles or drug delivery systems (e.g., liposomes, nanocarriers) for targeted delivery.
Advantages: The high selectivity and stability of click reactions are particularly beneficial in biological systems, minimizing off-target reactions and enabling the ASOs to maintain functionality.
Other Methods for ASO Conjugation:
In addition to NHS ester and click chemistry, other methods like maleimide-thiol coupling (for thiol-reactive conjugates) and carbodiimide coupling (EDC/NHS) are also used, but NHS ester and click chemistry are some of the most efficient and popular approaches due to their speed, versatility, and compatibility with various conjugates and environments.
Use in ASO Therapeutics:
Conjugating ASOs with targeting ligands (e.g., antibodies, aptamers) or delivery-enhancing agents (e.g., peptides, lipids) can improve their pharmacokinetics, reduce off-target effects, and increase their therapeutic efficacy. Both NHS ester chemistry and click chemistry are widely adopted in the biopharma industry for this purpose.
These methods are commonly employed in commercial ASO conjugation strategies to develop next-generation ASO therapeutics for improved targeting, efficacy, and safety profiles.
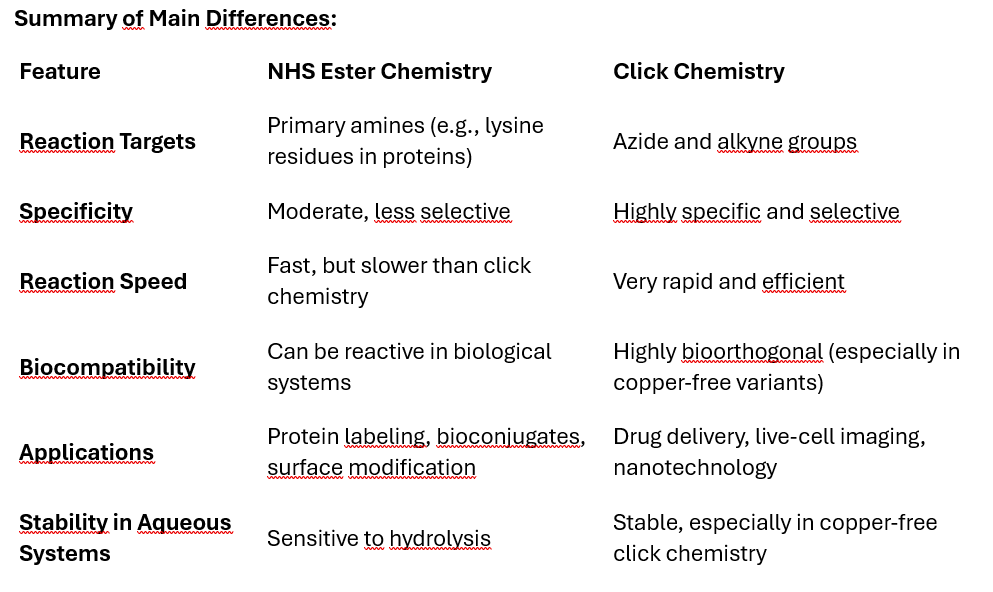
NHS (N-hydroxysuccinimide) ester chemistry and click chemistry are both commonly used methods in bioconjugation, but they differ in their mechanisms, specificity, efficiency, and applications. Here's a comparison of the two:
Mechanism of Reaction
NHS Ester Chemistry: NHS esters react with primary amines to form stable amide bonds. The amine group is usually present in proteins, peptides, or other biomolecules. The reaction takes place under mild conditions and requires a slightly basic pH (around 7.5-8.5).
Click Chemistry: The most common click chemistry is copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC), where an azide group reacts with an alkyne to form a triazole ring. This reaction is highly specific and efficient, often referred to as “bioorthogonal” because it doesn’t interfere with other functional groups present in biological systems.
Specificity
NHS Ester Chemistry: Less specific, as NHS esters can react with any primary amine group available. This could lead to off-target reactions or non-selective labeling in complex systems (e.g., proteins with multiple amine sites).
Click Chemistry: Extremely specific and selective, as it only occurs between azides and alkynes, making it highly useful for targeting specific groups without affecting other biomolecules. The bioorthogonal nature allows it to work in living systems with minimal side reactions.
Reaction Speed and Efficiency
NHS Ester Chemistry: NHS ester reactions are fast but not as fast as click chemistry. The reaction efficiency depends on the environment, such as pH, and can be reduced in the presence of competing amines (like those found in proteins or biological media).
Click Chemistry: Click chemistry is very rapid and highly efficient. The reaction typically proceeds to near completion without by-products, especially when catalyzed by copper (CuAAC).
Compatibility with Biological Systems
NHS Ester Chemistry: NHS esters are sensitive to hydrolysis, meaning they can degrade in aqueous environments, limiting their stability and shelf-life before use. Not ideal for in vivo (inside living organisms) applications due to the reactivity with multiple biological amines.
Click Chemistry: Click reactions (especially in CuAAC) can be performed in aqueous environments and are relatively inert to other biological molecules, making them more suitable for in vivo applications. Copper-free click chemistry variants (like strain-promoted azide-alkyne cycloaddition, SPAAC) are particularly useful for in vivo applications as they eliminate the need for potentially toxic copper catalysts.
Applications
NHS Ester Chemistry: Commonly used for protein labeling, peptide synthesis, surface functionalization, and creating bioconjugates by linking biomolecules via their amine groups.
Click Chemistry: Widely used in drug delivery, materials science, nanotechnology, and bioconjugation, especially in live-cell or in vivo studies due to its specificity and bioorthogonal nature. It’s also employed in creating complex molecules, attaching fluorophores for imaging, or attaching drugs to targeting ligands.
By-products and Purity
NHS Ester Chemistry: By-products include hydrolyzed NHS ester and other potential side products. Purification steps may be required to remove unwanted reactions.
Click Chemistry: Generates minimal by-products. The triazole formed in the click reaction is stable, and the reaction itself is typically cleaner.
Both methods have their advantages depending on the specific needs of the experiment or application, but click chemistry is often preferred for its precision and cleaner reactions in biological contexts.
Conclusion
The chemical modification of antisense oligonucleotides (ASOs) is a cornerstone of their development, critically enhancing their stability, specificity, and pharmacological properties. ASOs, in their unmodified state, are vulnerable to rapid enzymatic degradation, poor bioavailability, and potential off-target effects. However, by integrating precise chemical modifications into the backbone, sugar moieties, and terminal ends, these therapeutic molecules can be transformed into stable, highly specific agents capable of modulating gene expression effectively in vivo.
Backbone modifications, such as phosphorothioate (PS) linkages, represent one of the most widely used strategies for enhancing ASO performance. PS modifications increase nuclease resistance by replacing one of the non-bridging oxygen atoms in the phosphate backbone with sulfur, thereby reducing exonuclease and endonuclease affinity. This modification also enhances plasma protein binding, particularly to serum albumin, which extends the circulation time of the ASO and reduces renal clearance, thereby improving pharmacokinetics. However, the introduction of sulfur creates stereochemistry at each modified bond, leading to R and S diastereomers that can affect both the efficacy and potential toxicity of the ASO. This necessitates careful consideration of the chirality introduced during the modification process to balance stability with biological activity.
Sugar modifications at the 2' position of the ribose backbone are another key advancement in ASO chemistry. Modifications such as 2'-O-methyl (2'-OMe) and 2'-O-methoxyethyl (2'-MOE) enhance binding affinity to the target RNA by stabilizing the ASO-RNA duplex, leading to higher thermal stability and improved resistance to nucleases. Locked Nucleic Acids (LNAs) take this a step further by "locking" the ribose sugar into a rigid conformation, resulting in dramatic increases in binding affinity and target specificity. LNAs allow for shorter ASO sequences with higher potency and specificity, which can reduce off-target effects. However, the increased rigidity and affinity introduced by LNAs can sometimes lead to cytotoxicity, making it crucial to carefully optimize the number and position of LNA residues in the ASO to balance efficacy with safety.
End modifications are equally critical for improving the stability and longevity of ASOs in biological systems. The 3' and 5' ends of ASOs are particularly susceptible to exonuclease degradation, and capping these termini with chemical groups like inverted thymidine (InT), phosphate, or phosphorothioate significantly enhances ASO durability in vivo. Conjugation of ASOs with molecules such as polyethylene glycol (PEG) or cholesterol further improves pharmacokinetics by increasing solubility, reducing immune system recognition, and improving tissue distribution. These end modifications also enhance cellular uptake, especially in the case of cholesterol-conjugated ASOs, which exhibit improved membrane permeability and intracellular delivery.
Emerging modification strategies, such as peptide nucleic acids (PNAs) and tricyclic DNA (tcDNA), offer even greater stability and affinity enhancements. PNAs, for example, replace the natural sugar-phosphate backbone with a peptide-like structure, conferring extreme resistance to enzymatic degradation while maintaining high affinity for complementary RNA sequences. Similarly, tcDNA introduces tricyclic structures that not only improve binding affinity but also enhance specificity by reducing flexibility, which minimizes off-target interactions. While these modifications hold great promise for next-generation ASO therapies, they also present challenges, such as poor solubility and cell permeability, that must be addressed through innovative delivery strategies, including liposomes or nanoparticle systems.
In conclusion, chemical modifications are indispensable for the successful development of ASOs as therapeutics. Backbone, sugar, and end modifications work synergistically to optimize ASO pharmacokinetics, binding affinity, and stability, while minimizing off-target effects and immunogenicity. The precise integration of these modifications allows ASOs to become robust, clinically viable molecules capable of achieving targeted gene modulation with high specificity. As the field of ASO therapeutics continues to advance, emerging chemical strategies will push the boundaries of what is possible, enabling the development of next-generation ASOs that are not only more effective but also safer and more versatile. These advancements are poised to drive innovations in personalized medicine, offering new hope for treating a wide array of genetic disorders and complex diseases.
The next article will be part 4. Synthesis and Purification of ASOs, stay tuned..
Thanks for reading Biotechnology Reviews Journal! Subscribe for free to receive new posts and support my work.