

Building Better Libraries: Rational CDR Design, TRIM Oligos, and NGS Validation
Displaying millions/billions of unique antibody fragments on a genetic platform (phage, yeast, etc.) researchers can screen these libraries to targets

Antibody libraries are diverse collections of antibody variants used in in vitro selection platforms to discover binders with desired properties. By displaying millions to billions of unique antibody fragments on a genetic platform (phage, yeast, etc.), researchers can screen these libraries to isolate rare antibodies against virtually any target. The quality of an antibody library – in terms of its sequence diversity, functional expressibility, and representation of useful binding motifs – directly determines the success of downstream selection campaigns. This review provides a comprehensive overview of the technical and methodological aspects of antibody library design and diversity generation. We focus on the major library platforms (phage, yeast, and others) and the origin of their diversity (natural immune vs. synthetic libraries), strategies for maximizing functional diversity (rational design vs. random mutagenesis, structural constraints like CDR-H3 loops), and enabling technologies (advanced DNA synthesis, next-generation sequencing for library validation, and machine learning-guided design). We also discuss common challenges, such as construction biases, diversity bottlenecks, and the distinction between sequence diversity and functional diversity and highlight recent innovations from the past 3–5 years that are shaping the field. Throughout, the emphasis is strictly on technical and methodological considerations, rather than therapeutic applications.
Antibody Library Platforms and Formats
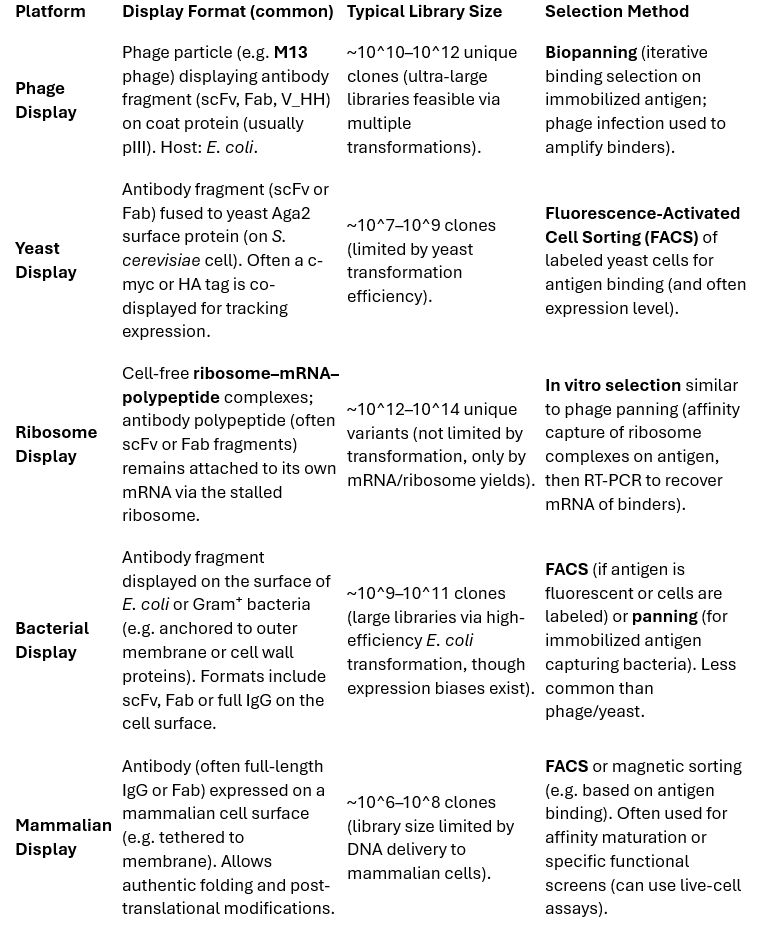
In vitro display technologies link antibody genotype to phenotype, allowing selection of binding clones from extremely large variant pools. The most widely used platforms are phage display and yeast surface display, with newer or specialized systems including cell-free displays (ribosome and mRNA display), bacterial display, and mammalian cell display. Table 1 summarizes key features of major library platforms.
· Library quality = success: Sequence diversity, functional expression, and useful binding motifs directly determine discovery outcomes.
· Multiple platforms: Phage and yeast dominate, but ribosome, mRNA, bacterial, and mammalian display expand options.
· Design matters: Smart diversity generation (rational design, CDR-H3 control, advanced DNA synthesis, NGS, and ML) drives modern library performance.
Table 1 – Comparison of Antibody Library Display Platforms (typical formats, library sizes, and screening methods):
Phage Display: Phage display was the first and remains the most widely used antibody library platform. Antibody genes (commonly as single-chain Fv or Fab fragments) are cloned into phage genomes or phagemids, fusing the antibody to a coat protein (usually pIII) so that each phage particle presents an antibody on its surface and carries the encoding DNA inside. Phage libraries can be made very large (tens of billions of unique clones) because filamentous bacteriophage infect E. coli with high efficiency. In practice, libraries up to ~10^11 10^12 have been reported by performing multiple high-efficiency electroporations. Selection is done by biopanning: the phage library is incubated with an immobilized or target-coated surface, unbound phage are washed away, and bound phage are eluted and amplified in bacteria. Iterative pannings enrich specific binders. Phage display is robust, relatively low-cost and cell-free (aside from E. coli propagation), and has yielded many antibodies. However, it can suffer biases: clones with slightly faster growth or display may dominate after multiple rounds (“clonal dominance”), possibly outcompeting higher-affinity binders. Additionally, phage are prokaryotic, so folding complex eukaryotic antibody formats (e.g. full IgGs) on phage is challenging (Fab or scFv fragments are typically used). New phage systems (e.g. using pIX or pVII coat proteins) and clever helper phage designs have been developed to improve display valency and phage propagation, but the monovalent pIII display remains standard. Overall, phage display allows extremely large library sizes and straightforward enrichment via binding, making it a workhorse of antibody discovery.
Massive scale: Phage display supports huge libraries (10¹¹ 10¹² variants) via efficient E. coli transformation.
Workhorse method: Robust, low-cost, and widely used for antibody discovery through iterative biopanning.
Limitations: Susceptible to clonal dominance and folding issues for complex eukaryotic antibody formats.
Yeast Display: Yeast surface display uses Saccharomyces cerevisiae to express antibody fragments on the cell wall, typically by fusing the V_H V_L fragment to Aga2, which forms a complex with the cell-wall protein Aga1. Each yeast cell displays many copies of the antibody fragment on its surface. Yeast libraries are constructed by transforming plasmid pools into yeast; library sizes around 10^7 10^9 are achievable, lower than phage libraries due to yeast’s more limited transformation efficiency (∼10^7 per μg DNA in optimal conditions). The major advantage of yeast display is the ability to directly screen for binding and other properties using flow cytometry. Yeast-displayed antibodies can be labeled with fluorescent antigens and an anti-tag or anti-Fab antibody to simultaneously measure antigen binding and expression level on each cell. Fluorescence-Activated Cell Sorting (FACS) can then gate for clones that both bind target and are well-expressed, enriching functional binders. This provides fine control for example, one can sort for clones that bind at pH 6 vs pH 7 to select pH-dependent binders, or isolate high-affinity clones by competition with a soluble antigen. Yeast being a eukaryote can perform correct disulfide bond formation and other folding processes, often yielding higher percentages of properly folded antibodies than phage. Another benefit is that poorly expressing or aggregation-prone clones tend not to display well and thus are implicitly deselected during FACS for binding vs. expression. On the downside, the library size is smaller and maintaining yeast libraries is more labor-intensive (requiring growth in culture). Yeast display is commonly used for affinity maturation and engineering of antibodies (starting from a smaller library of variants of an existing antibody), but it has also been used for de novo discovery from naïve or synthetic libraries.
Controlled screening: FACS enables simultaneous selection for binding and expression, enriching functional binders.
Eukaryotic advantage: Yeast ensures proper folding and disulfide bonds, filtering out unstable or poorly expressed clones.
Trade-offs: Library sizes are smaller (10⁷ 10⁹) and maintenance is more labor-intensive than phage display.
Ribosome and mRNA Display: These are cell-free display technologies that avoid any transformation into living cells, allowing enormous library diversities (theoretical >10^13). In ribosome display, a DNA library is transcribed and translated in vitro (e.g. in a reticulocyte lysate) under conditions that stall the ribosome at the end of the antibody fragment mRNA (often by lack of a stop codon). The result is a complex of mRNA, ribosome, and nascent antibody protein, where the antibody is physically linked to its mRNA via the ribosome. These complexes can be panned against an antigen similarly to phage. After binding and washing, the mRNA of bound complexes is recovered (usually by RT-PCR), and either analyzed or amplified for another selection round. Because no cell is involved, the library size is only limited by the DNA input and translation system on the order of 10^12 10^14 unique sequences can be screened in a single tube. A related method, mRNA display (e.g. the PET or SELEX variants), covalently links the nascent protein to its mRNA via a puromycin linker, achieving a similar genotype-phenotype coupling. These cell-free methods explore vast sequence spaces and allow incorporation of non-standard amino acids or unnatural chemistries if desired. Challenges include ensuring proper protein folding without cellular chaperones, and the instability of ribosome complexes. Nonetheless, they have been successfully used to isolate antibodies and scaffolds, and are a powerful complement to cell-based systems.
Ultra-large diversity: Enables screening of 10¹² 10¹⁴ variants without cellular transformation.
Cell-free flexibility: Allows incorporation of unnatural amino acids and chemistries.
Challenges: Protein folding is less reliable without chaperones, and ribosome/mRNA complexes can be unstable.
Bacterial and Mammalian Display: Other platforms have more specialized applications. Bacterial display can present antibodies on the surface of E. coli or other bacteria (for instance using autotransporters or cell wall anchors). This can achieve library sizes approaching 10^10 10^11 (since E. coli can be transformed almost as efficiently as in phage display). However, the complexity of displaying large antibodies on bacteria and issues like folding in the periplasm mean bacterial display is less common for general antibody discovery. It has found use in specific engineering tasks and epitope mapping. Mammalian display, by contrast, uses cultured mammalian cells (e.g. HEK293 or CHO cells) transfected with libraries encoding full-length IgG or Fab on the cell surface. The appeal is that full IgG molecules can be displayed and screened in a format very close to the final therapeutic form, with native folding and post-translational modifications. This can be crucial for complex targets (e.g. selecting antibodies that bind a cell-surface receptor in its native conformation). Mammalian display libraries have been smaller (~10^7 10^8 max) due to limits of DNA delivery and cell growth, but even libraries of 10^7 can yield hits for affinity maturation when combined with sensitive FACS sorting. Mammalian display is often used to refine lead antibodies (e.g. improve affinity or specificity) or to screen directly in cases where the highest fidelity of folding is needed (at the cost of throughput).
Bacterial display: High potential diversity (10¹⁰ 10¹¹) but limited by folding issues; mainly used for specialized engineering and epitope mapping.
Mammalian display: Presents full-length IgGs with native folding and modifications, ideal for complex targets.
Trade-off: Mammalian libraries are smaller (~10⁷ 10⁸) but deliver high-fidelity screening close to therapeutic format.
Each platform involves trade-offs between library size and screening precision. Phage and ribosome display offer enormous diversity but rely on in vitro or bacterial expression (which may include non-functional clones), whereas yeast and mammalian display offer more functional screening at the cost of library size. Increasingly, campaigns use multiple platforms in tandem for example, panning a huge phage library to get initial binders, then optimizing those binders by yeast or mammalian display. The choice of platform is a key technical decision in library design, with implications for how diversity can be generated and utilized.
Natural vs. Synthetic Antibody Libraries
Another fundamental distinction is the source of diversity in an antibody library whether the variability derives from natural immune repertoires or is introduced synthetically. Antibody libraries are often classified as natural (immune or naïve), fully synthetic, or semi-synthetic (hybrid). Each approach has technical advantages and challenges:
Natural Immune Libraries: These are libraries derived from real antibodies produced by B cells in vivo. In an immune library, B cells are obtained from an immunized animal or a human donor who has recovered from an infection (or been vaccinated), so that the repertoire is enriched for antibodies against a particular antigen. For example, after immunizing a llama with an antigen, one can clone the V_HH domains from its B cells into a phage library. Immune libraries often contain high-affinity binders to that specific antigen, thanks to in vivo affinity maturation. However, their diversity is inherently biased to that target; they are less universal and are used when one has a specific immunogen in mind. In contrast, naïve libraries are made from B cell repertoires of non-immunized donors (healthy individuals). Here, the diversity reflects the broad, baseline antibody repertoire that nature can generate without antigen-driven selection. Early landmark libraries, such as the Cambridge Antibody Technology (CAT) library, pooled IgM/G genes from dozens of healthy donors to reach >10^10 diversity. Naïve libraries aim to be universal (able to find binders to many different targets), but any given binder in a naïve library may be low-affinity since the donor’s B cells were never specifically matured for that antigen. The strength of natural libraries is that sequences are bona fide products of V(D)J recombination and somatic mutation they come with human-compatible frameworks and are more likely to fold and function. Natural libraries typically incorporate a wide variety of V gene families and paired heavy/light chains as they exist in nature (if cloning whole Fab or scFv from single B cells or combinatorially mixing donor heavy and light repertoires). This means they sample many framework structures and can present a broad array of CDR conformations seen in real antibodies.
Immune libraries: Built from immunized donors, enriched for high-affinity binders but biased toward the immunogen.
Naïve libraries: From non-immunized donors, broad and universal but often lower affinity.
Strength: Natural repertoires provide authentic V(D)J diversity, paired chains, and human-compatible frameworks.
Synthetic Libraries: A synthetic library is constructed by intentional design and mutation of antibody sequences in vitro, rather than directly using B-cell derived sequences. Typically, a small number of reliable frameworks (often human germline frameworks known for stability and “drug-like” behavior) are chosen as the scaffold. Then, diversity is introduced into the complementarity-determining regions (CDRs) by synthetic oligonucleotides during library cloning. Early synthetic libraries often used a single light and heavy chain framework (or a few) with entirely random sequences in one or more CDRs. For example, the Griffin library (MRC 1994) used dozens of human frameworks but fully randomized the heavy chain CDR3; later synthetic libraries like ETH2-Gold (2005) and HAL9/10 focused on one human framework with diversifications mostly in CDR-H3 and some in CDR-L3. The synthetic approach allows complete control over the diversity: one can incorporate any amino acid at any position, including unnatural patterns not readily found in nature. This is useful for targeting antigens where natural immune repertoires might be lacking. However, purely random diversification can produce many non-functional clones (sequences that do not fold or bind) indeed, an ongoing goal in synthetic library design is to maximize the fraction of “functional” antibodies among all the variants.
Designed diversity: Built on stable human germline scaffolds with CDRs diversified using synthetic oligos.
Control & flexibility: Enables precise amino acid choices, including motifs absent from natural repertoires.
Challenge: Purely random designs risk many non-functional clones, making functional enrichment a key goal.
Limitations of Traditional Degenerate Codon Libraries (NNK/NNS)
Conventional antibody library construction often relies on degenerate codons (e.g. NNK or NNS) to randomize amino acids, but this approach has inherent limitations. An NNK scheme produces 32 possible codons to encode 20 amino acids, introducing redundancy and typically a stop codon in ~3% of variants. This leads to many non-functional clones and an uneven amino acid distribution, as some residues are overrepresented due to codon bias [Wikipedia]. The random mixing of nucleotides in degenerate libraries also means designers cannot easily exclude undesirable codons or sequences. In practice, these factors force larger library sizes to achieve full coverage and can impede downstream screening efficiency. By contrast, next-generation methods have been developed to eliminate unwanted codons (e.g. stop codons or rare tRNAs) at the synthesis stage, improving the quality and usability of libraries [McLaughlin TRIM Tech] [Biotechnology Reviews].
TRIM Phosphoramidite Chemistry and Position-Specific Control Trinucleotide phosphoramidite (TRIM) technology addresses the shortcomings of degenerate oligos by using pre-formed codon building blocks in DNA synthesis. Instead of adding bases one by one, entire codons (triplets of nucleotides) are coupled as single synthetic units [McLaughlin TRIM A-Z] [Biotechnology Reviews]. This chemistry bypasses the randomness of traditional base-by-base coupling and avoids frame-shift errors, yielding higher-fidelity oligonucleotides for library construction [Biotechnology Reviews]. Critically, TRIM allows precise position-specific control: each variable position in the oligonucleotide can be programmed with a defined mix of codons, encoding a chosen subset of amino acids [McLaughlin TRIM A-Z] [Biotechnology Reviews]. This deliberate codon incorporation ensures the intended amino acids are represented without bias or unwanted codons [Biotechnology Reviews]. By encoding diversity at the codon level, TRIM libraries achieve accurate translation of designed diversity into protein libraries, with improved consistency and synthetic efficiency.
Codon Engineering: Per-Position Choices and Codon Optimization A key advantage of TRIM oligonucleotides is the ability to customize the genetic code at each randomized position. Library designers can specify per-position amino acid sets, for example omitting cysteine codons in a CDR to prevent disulfide artifacts or limiting a position to polar residues to preserve solubility. Unwanted codons (such as premature stops) are simply excluded from the codon mix, so no nonsense mutations occur in the library [McLaughlin TRIM A-Z]. Moreover, TRIM enables codon optimization tailored to the expression host’s tRNA biases. Codons can be selected to match the preferred usage in bacterial, yeast, or mammalian systems [McLaughlin TRIM Diversity] [Biotechnology Reviews], avoiding those that would slow translation in a given host. For example, an arginine codon like AGA might be favored for E. coli expression whereas rare alternatives are omitted [Biotechnology Reviews]. By engineering codon content per position, TRIM libraries maximize translational efficiency and ensure each position’s variability aligns with both functional needs and manufacturing constraints.
Eliminating PTM Motifs, Protease Sites, and Rare Codons Rational codon design further allows avoidance of sequence motifs that could impair protein display or function. One important consideration is eliminating post-translational modification (PTM) motifs. TRIM libraries can be designed to avoid introducing N-glycosylation sequons the N-X-S/T motif in antibody CDR regions [McLaughlin Codon/PTM] [ResearchGate]. This prevents clones from acquiring unintended glycosylation that could alter binding or be selected against. Similarly, codons can be chosen to ensure the library does not contain internal protease cleavage sites or other liability motifs (for instance, avoiding di-basic sequences that proteases recognize). Because TRIM synthesis excludes specified triplets, it inherently removes codons that are rarely used or problematic in the host organism’s context [McLaughlin Codon/PTM]. Rare codons that might reduce expression or accuracy in E. coli (e.g. certain Arg or Ile codons) are omitted up front [Biotechnology Reviews]. These design precautions yield libraries free of “hidden” pitfalls every variant is more likely to be expressed correctly and stably, without sequence features that could trigger degradation or modifications.
Codon-level precision: TRIM uses pre-formed trinucleotides to eliminate randomness, stop codons, and frame-shift errors.
Customizable diversity: Designers can tailor amino acid sets per position and optimize codons for the expression host.
Built-in quality: PTM motifs, protease sites, and rare codons are excluded, improving folding, expression, and stability.
Oligonucleotide Design: TRIM ssDNA Length, Doped Regions, and Mix Balancing The technical implementation of TRIM libraries involves careful oligonucleotide design to achieve the desired diversity. Typically, single-stranded DNA (ssDNA) cassettes containing randomized codon regions (e.g. diversified CDR loops of an antibody) are synthesized with lengths accommodating flanking constant regions for cloning. TRIM oligos often range from a few dozen bases up to ~150 bases, depending on the library size and vector context. Within these oligos, certain positions may be doped or partially randomized: for example, a position can retain the wild-type codon in a fraction of clones (to preserve function) while sampling alternatives at a defined frequency. Such doped regions are achieved by mixing a fixed codon with a low proportion of random codon during synthesis, allowing fine-tuned mutational spectra. Conversely, some segments can remain fixed (non-diversified) to maintain framework integrity. Another crucial aspect is mix ratio balancing for each TRIM codon mix. Because multiple codons can encode the same amino acid, chemists adjust the input proportions of each trinucleotide amidite to ensure uniform amino acid representation in the final library [Biotechnology Reviews]. This prevents over-representation of any residue and maintains an even diversity distribution. Overall, TRIM oligonucleotide synthesis provides flexibility in oligo length and composition, enabling complex library designs with controlled diversity and embedded structural constraints.
Library Construction Workflows: Uracil-Template Mutagenesis and Modular Assembly Once TRIM diversifiers are designed, they must be integrated into full-length antibody or protein library constructs. One common workflow is uracil-template mutagenesis (Kunkel mutagenesis), which is well-suited for phage display libraries. In this method, a single-stranded DNA template containing dU residues is generated, and the TRIM oligonucleotide (encoding diversified CDRs) is annealed to it as a primer. After extension and ligation, the heteroduplex is introduced into an E. coli repair strain that degrades the uracil-containing original strand, enriching for clones with the synthetic strand. This yields a library of phage or plasmids where the inserts carry the TRIM-designed mutations. An alternative or complementary approach is modular assembly using Golden Gate cloning (a Type IIs restriction enzyme method). In Golden Gate assembly, multiple dsDNA fragments which can include synthesized TRIM cassettes for each diversified region are ligated in a single reaction using designed overhangs. This strategy is powerful for assembling large synthetic antibody libraries from smaller DNA modules, or for combinatorially combining heavy and light chain repertoires. Golden Gate assembly ensures seamless joining of pieces (no scar sequences) and is highly amenable to building diverse libraries in a one-pot reaction. These workflows are often combined with PCR amplification steps to yield the final library, and each has distinct advantages in efficiency and diversity retention [McLaughlin DNA Lib]. Ultimately, TRIM oligonucleotides are compatible with both phage display (via Kunkel) and many plasmid-based library construction techniques (via modular cloning), giving researchers flexibility in how they generate the final library DNA.
Flexible design: TRIM ssDNA oligos (up to ~150 bases) can mix fixed, doped, and randomized codons, with balanced trinucleotide ratios for even amino acid representation.
Kunkel mutagenesis: Uses uracil-containing templates and TRIM primers to efficiently generate diversified phage display libraries.
Golden Gate assembly: Enables seamless, modular construction of large synthetic antibody libraries and combinatorial heavy/light repertoires.
Quality Control: Deep Sequencing and Library Quality Assessment Robust quality control is essential to validate that the constructed library meets design specifications. Deep sequencing (NGS) is routinely employed to sample the library’s diversity at the DNA level. This involves high-throughput sequencing of the diversified region from a portion of the library clones to verify the distribution of amino acids at each position. In a TRIM-based library, NGS should confirm that each intended amino acid frequency matches the design (e.g. equal representation if that was specified, or correct biased ratios) and that no unintended mutations or frame-shifts are present. It also allows estimation of clone diversity the number of unique variants by counting unique sequence reads, which helps gauge whether the library size approaches the theoretical diversity. Additionally, bioinformatic screening of the sequences can detect any forbidden motifs or codons that might have slipped through (for instance, confirming no clone contains a stop codon or an N-glycosylation motif if those were supposed to be eliminated). Beyond sequencing, other QC measures include analytical restriction digests or PCR of random clones to ensure insert length is correct, and occasionally functional assays on a small scale to make sure library members are expressible. Together, these quality control steps ensure the TRIM library is faithful to its design in terms of both the genetic diversity and the absence of deleterious sequences before it is used in downstream selection screens.
Advantages of TRIM Libraries over NNK Libraries By combining precise chemistry and rational design, TRIM libraries offer significant advantages compared to traditional NNK/NNS libraries. First, the functional diversity of a TRIM library is effectively higher every clone encodes an amino-acid sequence within the intended design space (no wasted clones carrying stops or irrelevant mutations). This means library size can be smaller yet cover the same functional sequence space, accelerating screens with fewer non-productive variants [Biotechnology Reviews]. Second, TRIM libraries are engineered for optimal expression fidelity. Codon choices are optimized to the host, and problematic sequences are removed, so cloned variants are more likely to fold and express correctly (e.g. in phage or yeast display) without biases from expression failures [McLaughlin Codon/PTM]. This leads to a more uniform pool of displayed proteins or antibodies, where differences in enrichment reflect binding properties rather than expression artifacts. Finally, these improvements translate to better downstream screening outcomes. Studies have noted that precisely diversified libraries increase the probability of finding high-affinity binders and specific functional hits, while reducing off-target or artifactual selections [McLaughlin TRIM A-Z] [Biotechnology Reviews]. In practice, lead antibodies from TRIM libraries tend to have fewer developability issues because the library was pre-filtered for expression-friendly features. Overall, TRIM oligonucleotide libraries empower researchers to explore protein sequence space with fine-grained control, yielding libraries that are not only diverse but also enriched in viable, expressible, and therapeutically relevant candidates. Such libraries markedly improve the efficiency of antibody discovery and protein engineering campaigns [McLaughlin TRIM A-Z] [Biotechnology Reviews], underscoring the value of codon-level precision in synthetic library design.
Higher functional diversity: Every TRIM clone encodes a valid amino acid sequence—no wasted variants with stops or frame errors.
Optimized expression: Host-specific codon usage and removal of problematic motifs improve folding and display fidelity.
Better outcomes: TRIM libraries yield more high-affinity, developable binders, boosting efficiency of antibody discovery.
Semi-synthetic libraries are a hybrid approach that tries to get the best of both worlds. A common tactic is to use natural sequences for the most critical regions (or to seed the diversity with natural motifs) while randomizing others. For example, third-generation “gold” libraries often keep heavy chain CDR3 loops taken from real human antibodies, while synthetic oligonucleotides diversify the other CDRs. The reasoning is that CDR-H3 is the most diverse in nature and crucial for antigen binding; by using a large collection of natural CDR-H3 sequences (e.g. cloned from human B cells) one captures realistic loop structures and avoids the astronomically large synthetic space of completely random H3. The remaining CDRs (L1, L2, L3, H1, H2) can then be diversified synthetically, but often in a constrained way (for instance, only allowing amino acids observed in those positions among natural antibodies, so as not to disrupt canonical loop structures). Many successful libraries (e.g. the HuCAL series, Althea’s libraries, etc.) are semisynthetic: they use human germline frameworks and apply synthetic diversity informed by natural antibody frequencies and motifs.
From a structural and functional diversity standpoint, natural vs. synthetic libraries can have different biases. Natural libraries have diversity encoded in a collection of discrete genes (the V, D, J segments and junctional modifications), so their diversity may be uneven (some motifs over-represented, others absent) and correlated (certain heavy-light pairings are common, others never occur). Synthetic libraries, in contrast, can sample “untainted” combinations (e.g. heavy chain from one germline with light chain from another, or unusual amino acids in certain CDR positions) this may yield novel binding modes, but also many sequences that nature avoided for good reason (e.g. they might fold poorly or be self-reactive). A notable observation is that many synthetic libraries rely predominantly on the heavy CDR3 for binding diversity, whereas natural antibodies often utilize multiple CDRs in concert. One study found that synthetic antibodies (with random CDR3s on a single scaffold) tended to make antigen contacts concentrated in that H3 loop, potentially limiting the range of epitopes recognized (so-called “effective diversity”). By contrast, natural antibodies frequently use canonical structures in CDR1 and CDR2 that contribute to binding in different ways. Modern synthetic designs have responded by incorporating more natural patterns: for example, using natural amino acid frequency profiles for each position, or explicitly grafting canonical loop sequences from databases into the library. The overall trend has been a convergence synthetic libraries are becoming more “natural” in their diversity encoding (to ensure functional folding), even as they push into new sequence space.
In practical terms, natural libraries often have higher initial hit rates (since many clones are naturally functional), whereas synthetic libraries require careful design to avoid a low functional hit rate. However, synthetic libraries can be engineered to avoid unwanted motifs (such as glycosylation sites or immunogenic sequences) and can focus on “drug-like” properties from the outset. Indeed, second-generation synthetic libraries explicitly removed problematic sequences (e.g. poly-reactive motifs, unstable frameworks) and optimized developability features. The choice between using a natural or synthetic library depends on availability of donor material, the target of interest, and intellectual property considerations (fully synthetic human libraries provide unique antibody sequences not directly isolated from donors). Many companies maintain large proprietary synthetic libraries, while others leverage immunization in transgenic mice (a form of immune library) both approaches have yielded approved therapeutic antibodies. From a design perspective, the key is that natural and synthetic diversity have complementary strengths, and engineering efforts often blend them to maximize functional diversity.
Hybrid strategy: Natural CDR-H3 loops are combined with synthetically diversified other CDRs, balancing realism with design flexibility.
Bias differences: Natural libraries reflect authentic but uneven gene usage, while synthetic ones explore novel combinations that risk misfolding or self-reactivity.
Converging designs: Modern synthetic libraries increasingly mimic natural patterns to boost functional hit rates and developability.
Strategies to Maximize Functional Diversity
Creating a high-quality antibody library is not just about sheer numbers of sequences it’s about ensuring a rich functional diversity: a wide range of properly folded antibodies with different potential binding modes. Achieving this requires deliberate strategies in library design. Two broad approaches are often balanced: random mutagenesis to generate vast sequence space, and rational design to bias the library toward likely-functional regions of that space. Additionally, the unique structural constraints of antibodies (especially the heavy-chain CDR3 loop) influence how diversity should be introduced.
Random vs. Rational Diversification: Early antibody libraries leaned heavily on random mutagenesis, introducing diversity with minimal human bias. This might involve using degenerate codons at CDR positions (e.g. NNK or NNS codons, which encode all 20 amino acids with one or a few stop codons) or error-prone PCR to randomly mutate residues. The upside is maximal exploration: truly novel sequences can emerge. The downside is that most random sequences will not fold or bind. Rational design uses knowledge of antibody structure and sequence preferences to guide diversity incorporation. For example, instead of randomizing an entire 10-residue loop with all amino acids, a rational design might allow only a subset of amino acids at each position perhaps based on frequencies observed in natural antibodies or contacts seen in known antibody-antigen structures. One common practice is to limit diversification to specific CDRs or even sub-regions of CDRs that are most likely to tolerate mutations. Heavy-chain CDR3 is often the sole or primary diversified region in synthetic libraries because it provides most antigen contacts in many antibodies and can vary in length and composition widely without disrupting the overall immunoglobulin fold. By contrast, framework regions and certain CDR positions critical for structural integrity are left unchanged to maintain stability and expression. Rational design can also involve incorporating structural motifs deliberately: for instance, designing a library to include a disulfide-bonded loop in CDR3 to mimic a camelid antibody’s convex paratope, or enforcing a β-turn motif in a CDR by appropriate residues. In silico modeling or data-driven approaches (discussed later) can help predict which variations are viable.
In practice, most modern libraries use semi-rational approaches: some positions (especially in CDR1,2 of heavy and light chains) may be varied according to a natural amino acid distribution rather than completely randomly. For example, one might allow mostly conservative mutations with a few wildcards at a given site, reflecting that site’s variability in known antibodies. This increases the likelihood that the antibodies fold and resemble real ones. Library designers also consider position coupling certain residues may be co-varied or restricted because they interact. Purely random libraries ignore such correlations (leading to many “self-conflicting” sequences that combine incompatible amino acids). More advanced libraries use defined sets of CDR sequences or computationally optimized combinations to ensure internally compatible mutations.
Another tactic to expand diversity is chain shuffling: independently diversify heavy and light chains and then pair them combinatorially. Simply shuffling existing V_H and V_L genes from different sources can create new specificities (as was done in some early semisynthetic libraries). On the synthetic side, one could create separate heavy chain libraries and light chain libraries and then randomly combine them in phage assembly to theoretically get a diversity equal to the product of both. However, not all heavy-light combinations will form functional antibodies. Some library constructions explicitly randomize heavy-light pairing (e.g. mixing heavy and light repertoires from different donors) to increase diversity beyond the sequence level this can yield novel pairings, but the risk is that many pairings might misfold or mismatch in binding kinetics. Typically, successful pairing requires at least germline compatibility (certain human V_H families prefer certain V_L families). Therefore, completely random pairing is less common; instead, designers may choose a few representative heavy frameworks and a few light frameworks known to pair broadly, and diversify within those sets.
CDR-H3 Loop Considerations: The heavy-chain third CDR (CDR-H3) warrants special mention because of its outsized role in antibody diversity. CDR-H3 is the most variable region in length and composition in natural antibodies, often penetrating deepest into antigen binding pockets. It is often critical for antigen specificity, and many synthetic libraries focus large diversity there. However, unconstrained CDR-H3 diversity is astronomically large for a 10-residue loop with 20 amino acids each, that’s 20^10 (10^13) possibilities, which far exceeds practical library sizes. Random CDR-H3 libraries will thus only ever sample a tiny fraction of possible sequences, and that sampling may be biased or incomplete (some sequences might not be made or maintained). One strategy is to limit the length range of CDR-H3 to focus on lengths that tend to be functional (e.g. 8 16 amino acids, excluding extremely long loops that might be unstable) Another is to introduce structured diversity in H3: for instance, include a pair of cysteines in some fraction of the clones to form a loop (common in many natural antibodies to constrain long H3 loops). Libraries like HuCAL explicitly designed CDR-H3 with cysteine loops at certain positions in some variants, achieving a mix of loop structures. Additionally, position-specific biases can be applied e.g. at the base of CDR-H3 (near the junction), glycine and small residues might be favored to allow tight turns, whereas mid-loop positions might allow more bulk or aromatic residues for potential binding contacts. Indeed, HuCAL-Platinum library design used a “loop length-dependent amino acid distribution” for CDR-H3, meaning the allowed amino acids were tuned depending on whether the loop was short or long. All these measures aim to ensure that the library covers a variety of H3 loop conformations (length and shape) with amino acids that are conducive to proper folding and antigen recognition.
Summary of Diversity Design Tactics: To maximize functional diversity, library creators commonly:
Choose favorable frameworks: Use human germline or consensus frameworks known to be stable and free of liabilities. This reduces the chance that diversified clones will misfold.
Target the right regions: Focus randomization on CDRs (especially heavy chain CDR3, and often light chain CDR3 and heavy CDR2) while keeping frameworks and structurally crucial residues constant. This concentrates diversity where it most affects binding, not stability.
Use smart degeneracy: Instead of NNN (which includes 33% stop/non-functional codons), use NNK or NNS codons (encode 20 amino acids + 1 stop) or specialized codon mixes that exclude stop codons and under-represent cysteine (to avoid unintended disulfides). Many libraries use NNK (where K = G/T) which yields 32 codons including one stop (~3% of clones) some use NNS (S = G/C) to further reduce stop frequency. Alternatively, use defined mixtures of codons (trinucleotide phosphoramidites) to precisely encode allowed amino acids.
Bias toward natural patterns: Incorporate amino acid frequency profiles observed in natural antibody repertoires for each CDR position. For example, position 52 in heavy chain often is tyrosine or serine in real antibodies a synthetic library might allow Tyr/Ser at high frequency and a smattering of other residues instead of a flat random mix. This improves the chance of retaining canonical structures.
Avoid incompatible sequences: Omit or minimize combinations known to be problematic. E.g., avoid glycine at positions that require a β-carbon for structural reasons, or avoid adjacent prolines in a CDR which might rigidify it too much. If two positions are structurally coupled, don’t randomize both freely either co-design them or fix one. Such coupling can also be addressed by multi-step library building (first randomize some positions and select functional partial variants, then randomize the next set).
Remove liabilities: Ensure motifs that could cause trouble are not introduced. For instance, N-linked glycosylation sites (N-X-S/T) in CDRs might be removed from design (unless intentionally desired). Likewise, avoid motifs that trigger unfolded protein response or protease cleavage in yeast, etc. Modern libraries often explicitly eliminate sequences with potential PTM sites, protease sites, or aggregation-prone patches.
Validate in pieces: An advanced technique is to validate parts of the library in isolation. For example, one can construct sub-libraries for each CDR and test if those variants are well-expressed or bind a model target. In a published “giga-library” approach, researchers diversified all six CDRs but first subjected each diversified CDR (inserted into the scaffold one at a time) to a selection for proper folding using a β-lactamase enzyme fusion. Only variants of each CDR that passed the folding/activity filter were then combined to build the final library, drastically enriching the functional fraction. Similarly, MorphoSys’s HuCAL used a β-lactamase-based genetic selection to eliminate frameshifted or nonfunctional clones during library assembly. These quality-control steps at the design stage can greatly improve library functionality.
By carefully balancing random exploration with rational constraints, library designers aim to cover the broadest swath of functional sequence space. The ultimate measure of success is the percentage of library clones that can yield a binder to some antigen. A poorly designed library might have only 1% functional clones, whereas a state-of-the-art library might have >50% of clones properly folded and a high likelihood of finding sub-nanomolar binders to many targets. In the next sections, we delve into the technologies that enable these designs specifically, how synthetic DNA techniques and high-throughput sequencing are used to create and validate such diverse libraries.
Oligonucleotide Synthesis and Library Construction Techniques
The construction of a synthetic or semisynthetic antibody library hinges on DNA engineering: one must introduce the desired mutations and diversity into antibody gene sequences at specific positions. Several technological advances in oligonucleotide synthesis and cloning have made it possible to build libraries with precise control over diversity.
Degenerate Codon Mutagenesis: The simplest method to generate diversity is to use degenerate oligonucleotides during PCR or gene synthesis. For example, to randomize a CDR of length 5, one can design a primer encoding NNK at those positions (N = A/T/G/C, K = G/T). During oligo synthesis, each N position is a mixture of the four bases, so the primer pool contains a mix of sequences encoding all 20 amino acids (plus one stop codon) at those 5 positions. This pool is then PCR-amplified and cloned into the antibody framework vector, creating a library. Degenerate codons like NNK or NNS are standard because they minimize stop codons. Some libraries have used NNY (Y = C/T) or other combinations to exclude stops entirely. While straightforward, this approach has limitations: (1) unequal amino acid representation e.g. NNK gives 32 codons for 20 amino acids, so some amino acids (encoded by 1 codon) are underrepresented 3-fold compared to others (encoded by 3 codons). (2) Sequence bias DNA synthesis is not perfectly uniform; certain codon mixtures might favor AT-rich sequences, etc., leading to underrepresentation of some variants. (3) Coupling of mutations if many positions are randomized at once with degenerate primers, each clone picks a random combination, many of which may be incompatible as discussed. Nonetheless, degenerate codon mutagenesis is quick and has been used in countless libraries, especially when diversifying relatively few positions (such as affinity maturation libraries where maybe 5 10 positions around an antibody’s antigen site are targeted).
Trinucleotide Mixes (Codon-based diversification): To address the amino-acid distribution issue, methods were developed to synthesize oligonucleotides using pre-formed trinucleotide phosphoramidites, each corresponding to a codon. This “codon-based” synthesis (commercialized as TRIM technology by companies like Entelechon/SLS and used in later HuCAL libraries) means the library designer can specify exactly which codons (hence which amino acids) and at what frequency to incorporate at each position. For instance, if one wants 5% cysteine, 0% stop, and no arginine (to avoid isoelectric point issues) at a given position, one can mix the appropriate codons in those proportions. This yields a much more controlled library, often aiming to mimic natural amino acid frequencies. Moreover, codons can be chosen for expression optimality (avoiding rare codons that might impair translation in E. coli), which can increase the functional library size. The downside is that making and handling trinucleotide mixtures is complex and historically expensive, but it has become more accessible and several high-quality libraries have used this approach to eliminate biases and stops (e.g., HuCAL-GOLD and PLATINUM libraries).
Gene Assembly from Synthetic Oligo Pools: Traditional degenerate PCR methods are limited in the number of positions you can randomize simultaneously (too many degeneracies and the PCR won’t amplify well or the library size needed becomes impractical). A revolutionary advance has been the ability to synthesize large pools of designed oligonucleotides (100 200 bases or longer) on microarrays or other multiplexed synthesizers. Companies like Agilent (SurePrint technology) and Twist Bioscience (silicon-based synthesis) can produce pools containing thousands to hundreds of thousands of unique DNA sequences in parallel. Library designers can leverage this by encoding each variant (or segments of variants) explicitly rather than by degenerate codes. For example, one could design 10,000 distinct CDR3 sequences, each specified in an oligonucleotide, covering a carefully curated diversity of lengths and motifs. These olos can then be amplified and inserted en masse into the antibody scaffold. Because each sequence is defined, one avoids the combinatorial explosion of purely degenerate libraries essentially you sample a defined subset of sequence space. High-throughput oligo synthesis has allowed the creation of “digital” libraries where every clone’s sequence is predetermined (or at least drawn from a predetermined set). This enables incorporation of advanced design criteria: e.g. ensure each CDR variant appears once, exclude any with motifs one dislikes, include many known functional CDRs from databases, and so on. One successful example was a “pre-defined CDR” library where ~4000 actual CDR sequences (extracted from known antibodies sharing the same framework) were synthesized and assembled into a single framework. This yielded a library of >10^10 with each CDR known to be functional in that context, giving a >20-fold increase in unique hits during panning compared to a standard degenerate library. Such approaches are extremely promising for raising the baseline quality of libraries.
The assembly of full antibody genes from pools of oligonucleotides can be done by methods like overlap extension PCR, Golden Gate cloning, or Gibson assembly. Golden Gate assembly is particularly useful for libraries because it can seamlessly join multiple fragments in one pot using Type IIS restriction enzymes. For instance, one can synthesize heavy chain CDR3 variants as short fragments with flanking sequences that dictate assembly into the heavy chain gene via Golden Gate. Multiple positions (CDRs) can be simultaneously varied by mixing pools of fragments. The Golden Gate approach avoids intermediate cloning steps that could bottleneck diversity (since everything ligates in one reaction). Careful design of fragment overlaps and ratios is needed to ensure even representation, but when done right, it helps maximize the combinatorial assembly of diversity without bias.
Kunkel Mutagenesis and Other Techniques: A classical method in phage library construction is Kunkel mutagenesis, which uses an M13 phage gapped DNA and a uracil-containing template to incorporate oligonucleotide mutations site-specifically. This technique was used by some groups to introduce diversity into phage display vectors without needing PCR for the entire gene (thus potentially reducing size bias). Essentially, one produces single-stranded DNA with uracil, anneals a mixture of mutagenic oligonucleotides (with degenerate sequences) to it, extends and transforms into E. coli, which repairs and creates a library. Kunkel’s method yields high mutagenesis efficiency and was employed in several synthetic libraries (e.g., Griffin, HAL) to randomize CDRs.
Another approach to generate diversity is DNA shuffling or mixing of natural segments. For example, the nCoDeR library (BioInvent) took the approach of cutting human antibody gene segments corresponding to CDRs and recombining them into single frameworks. It essentially “shuffled” natural diversity into a new context. While not random at the nucleotide level, this combinatorial reassembly can create novel antibodies that are mosaic of natural parts.
Library Size and Transformation Efficiency: A practical consideration in library construction is ensuring the physical library size (number of independent clones) is sufficient to cover the intended diversity. If one designs 10^10 unique DNA sequences but only transforms 10^8 bacteria, then ~99% of designed sequences never make it into the library. It’s therefore critical to optimize transformation protocols and cloning yields. For phage display, electroporation of E. coli is typically used; with very high efficiency cells one can often get >10^9 clones per 0.1 1 μg of DNA. By scaling up (multiple cuvettes, lots of DNA), libraries of >10^10 can be achieved. Yeast, as mentioned, is harder to transform at scale methods like high-efficiency electroporation, lithium acetate/PEG, or cell fusion have been explored to push yeast library sizes towards the 10^9 range. In any case, one often needs to perform library size QC plating dilutions to count colonies or using NGS to estimate unique clone counts to confirm the library size. Sometimes, researchers will construct sub-libraries and then pool them to increase total diversity (ensuring each sub-library explores a different sequence space so there’s less redundancy).
In summary, modern oligonucleotide synthesis and cloning techniques give library designers an unprecedented level of control. We can now write DNA with huge complexity and defined composition, rather than relying on crude random mutagenesis alone. This has led to libraries that are not only large but also finely tuned for instance, libraries that precisely mimic the human repertoire’s germline usage and CDR diversity, or libraries biased toward certain structural solutions (like long-loop binders). As these synthetic biology tools continue to improve (cheaper DNA synthesis, better assembly methods), we expect even more creative library designs to emerge, pushing the envelope of functional diversity.
Next-Generation Sequencing for Library Validation and Evolution
The introduction of next-generation sequencing (NGS) has been a game-changer for antibody library analysis. In the past, one could only sequence on the order of 100 clones (via Sanger sequencing) to get a glimpse of library content. NGS now allows deep sequencing of millions of clones from a library, providing a detailed picture of its diversity and any biases. This has multiple important applications:
Library Quality Control: After constructing a library, researchers often perform NGS on the unselected library to verify that the diversity matches the design. For example, if a synthetic library was designed to have no tryptophan in CDR-L1 and 5% cysteine in CDR-H2, deep sequencing can confirm these frequency distributions. NGS can reveal if certain positions deviated from intended frequencies (due to synthesis or PCR bias) and identify over-represented or under-represented sequences. It’s not uncommon to find that, say, 1000 clones in a “random” library were actually identical indicating a PCR jackpot or clonal amplification event. With NGS, such issues are caught early and can sometimes be remedied (for instance, by re-synthesizing that part of the library or spiking in under-represented variants). NGS can also estimate the unique library size: by sequencing enough reads to see when they saturate unique variants, one can extrapolate how many unique clones the library contains. If a library was aiming for 10^10 but NGS indicates only 10^8 unique sequences, something went wrong (likely a bottleneck in cloning).
Bias and Diversity Metrics: NGS data allows calculation of diversity metrics like Shannon entropy at each randomized position, clonotype diversity indices, and other statistics to quantify how even or skewed the library is. For example, one can report the effective number of amino acids at each position (if some amino acids dominate, the effective diversity is lower). If an ostensibly random library shows very non-uniform distributions, that might prompt redesign. Importantly, NGS is often done without PCR amplification (PCR-free library prep) to avoid distortion of clone frequencies, since PCR can over-amplify some sequences. Protocols now exist to sequence antibody libraries by directly ligating adaptors and using unique molecular identifiers (UMIs) to correct any amplification bias.
Tracking Selection Rounds: Perhaps the most powerful use of NGS is to monitor how the library composition changes during the panning or sorting process. By sequencing the output of each round of phage panning, for instance, one can see which clones are rising in frequency and which are dropping. This can guide decisions like how many rounds to do (if a few clones have already dominated by round 3, additional rounds might only amplify those further, risking loss of others). Moreover, NGS reveals binders that might be missed by traditional picks in phage panning, one typically would pick 10 20 plaques to Sanger sequence and might end up with a few copies of the top clone. But NGS can show hundreds of distinct binders at lower frequencies. Some of those rarer sequences might actually be high-affinity or unique epitope binders that simply didn’t amplify as well. Researchers now often use NGS after 2 3 rounds to identify dozens of candidates, rather than just picking the most frequent. This greatly increases the chance of finding diverse binders (not just clonal siblings). In yeast display, similarly, one can sequence the library before and after a FACS sort to see enrichment. There are even strategies where one performs deep sequencing after a single sort or a single round, and then uses computational analysis to choose candidates without further laborious rounds.
Clonal clustering and lineage analysis: For immune libraries or affinity maturation libraries, NGS enables analysis of clonal families and maturation pathways. By clustering sequences by CDR-H3 similarity, one can group the outputs of selections into families that likely derive from the same precursor. This is useful to understand if a selection is yielding many unique solutions or just variants of one solution. If a dozen sequences differ only by a few residues, they might be affinity-improved mutants of one original binder. If another cluster is totally unrelated in sequence, that’s a distinct binder possibly hitting a different epitope. Such analysis of H3 clusters (or full VH-VL clusters) guides the selection of a diverse panel of hits for further characterization. It also allows one to measure effective diversity of hits: for example, maybe 1000 sequences after panning boil down to 5 clusters, indicating 5 distinct binders. If those were all sequence-unique one might have overestimated the diversity of outcomes.
Repertoire profiling and ML training data: The large datasets from NGS can feed machine learning models (as discussed in the next section). But even without ML, they give insight into selection stringency. For instance, one can calculate enrichment ratios of each clone between rounds, identifying which clones had the highest gains (likely good binders). One can also search within NGS data for specific motifs or germline usages to see if the selection is biased (e.g. does one heavy germline dominate the output?). If a certain framework is over-represented, it could mean that framework had an intrinsic display or growth advantage.
NGS in library evolution: There’s an emerging paradigm of using NGS iteratively to refine libraries. One can create a library, pan it, sequence it, and from the sequencing information design a second-generation library that fills in gaps or eliminates biases observed in the first. For example, if NGS shows that in the output binders there was an overwhelming bias for a certain motif, one might engineer the next library to either focus on that motif (if it seems to confer binding) or do the opposite (to explore other motifs that were neglected). Some researchers have also started using deep mutational scanning on antibody fragments systematically mutating positions and using NGS to measure which mutations are tolerated (via a binding selection) to map out where an antibody can tolerate diversity. This information can guide library design for affinity maturation (only mutate positions that don’t abrogate function).
In summary, NGS has become an indispensable analytical tool in antibody library technology. It moves the process from a black box to a data-driven exercise: one can debug and rationalize the library composition and selection progress. The cost of sequencing has dropped such that even academic labs routinely sequence their libraries and outputs. The result is higher fidelity libraries (since we can confirm they match design) and more efficient discovery (since we can harness many binders from a single campaign by sequencing). As library sizes continue to grow, NGS is the only feasible way to characterize millions of clones it essentially acts as a high-throughput analysis pipeline that complements the experimental screening.
Machine Learning and Computational Diversity Design
In the past 5 years, the field of antibody engineering has seen a surge in machine learning (ML) and AI-driven approaches that complement laboratory library methods. These computational techniques are being used to model antibody sequence-function relationships and even to design new library variants in silico. The goal is to guide diversity generation toward sequences with a higher chance of success (binding, stability, etc.), effectively improving the efficiency of library screening. Here we discuss how ML is interfacing with antibody library design:
One major use of ML is in analyzing large datasets (often obtained from deep sequencing or high-throughput assays) to learn what makes an antibody functional. For example, given an NGS dataset of binders vs. non-binders from a panning experiment, one can train a classifier model that distinguishes sequences that bound the antigen from those that did notFeatures for such models can include amino acid identity at each position, biochemical properties, or even computed structure features. Modern approaches often use protein language models or deep learning that take the raw sequence and automatically derive an informative representation. The trained model can then predict for new sequences whether they are likely to be functional binders or not. This provides a powerful in silico filter: instead of blindly including all variants, one could score candidate library members with the ML model and select only those above some predicted fitness threshold. A recent study demonstrated this by using a Bayesian optimization and transformer-based language model to generate new scFv sequences optimized for binding a target. They showed that an ML-designed library yielded a dramatically higher fraction of improvements (99% of variants improved binding over the starting antibody, whereas a conventional mutagenesis library had far fewer). This highlights how ML can explore sequence space in a directed way, finding beneficial mutations that might be missed by random mutagenesis.
Another application is generative models for antibody diversity. Techniques like generative adversarial networks (GANs) or variational autoencoders (VAEs) and, more recently, diffusion models have been used to generate novel antibody CDR sequences with certain desired properties. For instance, the AB-Gen framework used a generative pretrained transformer (GPT) with reinforcement learning to create CDR-H3 sequences that satisfy multiple property constraints (e.g. binding to a particular epitope as predicted by docking, plus good developability scores). By generating hundreds or thousands of such sequences in silico, one can synthesize a focused library that is highly enriched in potentially functional antibodies. These models are trained on massive databases of known antibody sequences (and sometimes structures), thereby learning the complex rules of antibody architecture. As a result, the sequences they generate often “look” like real antibodies, respecting things like canonical loop structures and conserved motifs, which naive random sequences might violate.
ML for affinity maturation is another area: given a specific lead antibody, one can use ML to suggest the best set of mutations to improve affinity or other traits. Instead of a researcher deciding which residues to mutate and in what combinations, algorithms can predict which positions are most likely to increase binding if mutated. These predictions can be used to design smaller, smarter libraries (e.g. focusing on 5 positions with top-scoring mutations rather than 50 positions randomly). There are now examples where such ML-guided libraries yielded improved binders in one or two rounds, whereas traditional methods might require many iterative rounds of mutagenesis.
Importantly, ML models can also incorporate developability and biophysical properties into the design criteria. Antibodies need more than just high affinity; they require good stability, low aggregation, low immunogenicity, etc. Researchers have started training ML models to predict properties like aggregation propensity, expression yield, thermal stability (T_m), and even propensity for specific liabilities (like polyreactivity). By scoring sequences on these traits, one can steer the diversity to avoid problematic regions. For example, one might use an ML model to eliminate any sequence that is predicted to have a low stability or a likely unfoldable region, even if that sequence might bind well. This integration of multiple objectives is where ML shines it can find sequences that balance affinity with other properties (a multi-objective optimization), something that is very hard to do manually or with single-property directed evolution.
One notable trend is the idea of in silico library screening: simulating or predicting the outcome of a library selection without doing it physically. If one has a good model of what makes a binder, one could in principle take a virtual library of millions of sequences, score them, and pick the top 0.1% to actually synthesize and test. This is analogous to virtual screening in small-molecule drug discovery. While not yet perfect, such approaches are improving. For instance, one group generated a virtual library of mutations on an antibody and used an ML model to predict their binding scores, choosing a subset to experimentally test; they found novel high-affinity mutants that traditional methods hadn’t found. As these predictive models become more accurate (benefiting from large public datasets of antibody sequences and some functional data), they could greatly reduce the experimental library sizes needed focusing wet-lab effort on sequences with high prior probability of success.
Machine learning can also assist in library analysis for example, using unsupervised learning on sequence data to identify clusters or patterns that correlate with function, or using language models to generate an “embedding” of antibody sequences in a continuous space. In that space, one could interpolate between known binders to find new ones, or determine if a library covers regions of natural repertoire space sufficiently. In fact, dimensionality reduction (like t-SNE or UMAP) applied to sequence embeddings has been used to visualize what subspace of sequence space a library or selection covers. Such analyses have shown that ML-optimized libraries can explore sequence areas not covered by conventional libraries, expanding the search space.
It’s worth mentioning that computational design doesn’t replace experimental libraries and selection rather, it augments them. Even the best prediction models have uncertainty, and there are always unmodeled effects (like how a given sequence actually folds in the context of the whole antibody). Therefore, current ML-guided designs still result in libraries or sets of candidates that need to be tested in the lab. But they can cut down the library size or increase the hit rate, making the experiments much more efficient. The synergy of high-throughput data (NGS, deep mutational scans) and ML is a virtuous cycle: data trains better models, and better models inform better library designs, which in turn yield better data.
In the coming years, we can expect ML to take on a larger role in de novo antibody design possibly generating full-length antibodies specific for an antigen from scratch (by combining structural modeling with sequence generation). Already, there are examples of designing antibodies to bind a given epitope on an antigen using computational methods (though not purely ML-based) for instance, by grafting CDRs predicted to fit an epitope and then optimizing. Fully ML-driven approaches might involve models that given an antigen or a paratope specification, propose antibody sequences likely to bind. Those would essentially bypass the random library stage, instead producing a focused set of candidates. However, for now, combinatorial libraries remain indispensable, and ML mainly helps to guide the composition of those libraries and interpret the results.
Challenges and Pitfalls in Library Design
Despite the tremendous progress in methods and scale, antibody library generation faces several persistent challenges that researchers must account for:
Biases in Library Construction: At every step of library creation, biases can creep in, skewing the diversity away from what was intended. During oligonucleotide synthesis, some sequences may be synthesized more efficiently than others (e.g., high-GC or repetitive sequences might have lower yield), resulting in their under-representation. PCR amplification can also introduce bias for instance, certain sequences may amplify faster due to secondary structure or better primer annealing, leading to over-amplification of those and dilution of others. Cloning and transformation into E. coli can impose bias: shorter or simpler inserts often transform more efficiently, so if a library contains a mix of insert sizes (e.g. varying CDR-H3 lengths), one might observe over-representation of shorter inserts in the final library. Indeed, special techniques (like length normalization or two-step cloning) are sometimes used to ensure equal representation of different CDR length classes. Additionally, during phage packaging, phagemid vectors with certain sequences might produce phage less efficiently (perhaps due to toxicity of the peptide or slower growth). These subtle biases mean the actual library can diverge from the theoretical design. NGS validation, as discussed, is crucial to detect these issues. When biases are found, one might take corrective measures, such as rebalancing codon mixes or performing additional rounds of diversification on under-represented regions.
Diversity Bottlenecks: A “bottleneck” refers to any step that drastically limits diversity. The most obvious is the transformation step if you only get 10^8 clones out of a theoretically diverse pool of 10^10, you’ve bottlenecked to 1% of the intended diversity. Another bottleneck can occur in panning: for example, after a very stringent wash, you might elute only 10^4 phage; if those came from 100 unique binders, that’s a severe squeeze of diversity from the original 10^10. Overly harsh selection or too few output clones can cause you to miss many binders (they may have been in the library but did not survive the bottleneck). Multi-round panning amplifies bottleneck effects by round 3, often a few clones dominate the pool, not necessarily because they are the best binders, but sometimes because they had a slight edge or just a lucky start. To mitigate this, some protocols use parallel panning strategies (multiple selections in parallel with different conditions) or limit the number of rounds and instead rely on NGS to pick out binders without over-amplification. In yeast display, bottlenecks occur if one gates too tightly in FACS (taking only the very top 0.1% might focus only on a few clones). A gentler initial sort followed by a tighter sort can help preserve more unique variants.
Functional vs. Sequence Diversity: Not all sequence diversity translates to functional diversity. A library could have a million unique sequences, but if 90% of them misfold or are not expressed, the effective library size is only 100,000 functional variants. This is why the percentage of functional clones is a key metric. Functional diversity also relates to the variety of binding solutions present. It’s possible to have many unique sequences that all bind the same epitope on an antigen essentially they represent one solution (maybe all using a similar H3 loop conformation). Another set of unique sequences might bind a different epitope, representing a second functional solution. For therapeutic discovery, one often wants multiple epitope binders, so the library needs to allow that. Synthetic libraries that focus too narrowly (e.g. one framework, one loop shape) might give many sequence-different hits that all compete for the same epitope on the target. Natural immune libraries might have biases to immunodominant epitopes (the immune system often focuses on one part of the antigen). This challenge can be tackled by designing libraries to include multiple structural frameworks (which may present different paratope shapes) and by performing selections in ways that promote diverse outcomes (for example, using competitive elution with a known binder to force discovery of binders to other epitopes). It’s also important to assay functional diversity in outputs e.g., binning binders by competition or mapping to ensure you didn’t just get redundancy.
Developability and Expression Issues: A library might yield binders that are technically functional (they bind the target) but are hard to produce or unstable, especially if the library didn’t consider developability. This is more a challenge in library usage, but it feeds back to design. Third-generation libraries have addressed this by building in developability selection (e.g. heat shock step to eliminate unstable clones, or using mammalian display to ensure clones express well in that system). If a library is generating many hits that later fail in expression, it might indicate that the diversity included some problematic region (like hydrophobic patches or certain germline sequences) redesign might be needed to excise those.
Intellectual Property (IP) and Redundancy: From a practical standpoint, if a synthetic library uses exclusively human sequences or known CDRs from literature, the hits you get might overlap with known antibodies (raising IP issues) or might be skewed toward well-explored sequence space. Some diversity generation efforts actually consider the novelty of sequences (trying to stay in human-like space but not identical to any known antibody). This is a softer challenge, but relevant for industrial libraries.
Screening Strategy Mismatch: A technical pitfall can occur if the library format isn’t optimal for the screening method. For example, using a Fab library in phage display but then trying to sort by FACS (which generally works better with cell-displayed formats) could limit the ability to directly screen on certain functional assays. Each library format may have sweet spots of what can be screened (phage is great for binding; yeast can allow screening for binding at different pH or even for enzymatic inhibition in some cases by labeling accordingly; mammalian can screen for cell signaling function, etc.). If one needs to screen for a complex function (like blocking a receptor in situ), a phage library might have to be converted to IgGs and tested in cell assays, which is a multi-step process and a potential bottleneck. This consideration can influence library design e.g., one might design a smaller mammalian display library specifically to allow direct functional screening in a cell-based assay.
Addressing these challenges often requires a multifaceted approach: careful design (to maximize functional fraction and ensure needed diversity), thorough validation (using NGS and small-scale expressions), and sometimes iterative improvement. Many groups iterate library designs the first generation teaches where the biases and holes are, the second generation fixes them, and so on. Indeed, the progression from first-gen to second-gen to third-gen phage libraries in the field exemplifies learning from challenges: first-gen had size but many non-functional clones, second-gen introduced natural distributions and removed obvious liabilities, third-gen added folding selections and developability screening. The result has been steadily improving library performance.
Recent Trends and Innovations (2020-2025)
The past 3-5 years have seen several exciting trends in antibody library technology, driven by both improved experimental methods and computational tools:
Integration of AI/ML in Library Design: As discussed, machine learning is now being actively used to design antibody libraries or guide affinity maturation. Approaches like generative models (including very recent diffusion models) are providing ways to explore sequence space efficiently. Notably, in 2022 2023 there were multiple reports of ML-designed libraries that achieved higher affinity improvements than traditional methods. Companies and academic teams are developing AI platforms to predict “hit-rich” regions of antibody sequence space before actually making the library, which could dramatically reduce trial-and-error in library creation.
Ultra-large Synthetic Libraries: While billion-scale libraries have been around, recent work has pushed the envelope further with giga-scale libraries constructed with high precision. For instance, a 2023 study reported a synthetic human scFv library of 2.5 × 10^10 clones built on a single human scaffold with six diversified CDRs. Key to this was the use of robust DNA synthesis and a novel assembly approach: each CDR diversity was first vetted by a functional selection, then combined, akin to assembling validated “building blocks”. The result was a library yielding sub-nanomolar antibodies with good biophysical properties. This demonstrates an innovation trend of quality filtering at construction making huge libraries but not sacrificing quality for size. Also, DNA synthesis companies (like Twist, Agilent) continue to increase the length and number of oligonucleotides they can synthesize in parallel, enabling more ambitious library designs. We now see libraries with tens of thousands of precisely defined CDR variants (covering, say, every possible combination of certain key residues) which were infeasible a few years ago.
Developability-Focused Libraries: A major theme of the new “third-generation” libraries is incorporating developability and here-and-now functionality into the library, not just after the fact. This includes: using only germline frameworks that have been proven in therapeutics (to avoid odd ones that might be problematic); engineering leader sequences or codon usage for high expression; eliminating sequence motifs that could cause aggregation or PTMs (e.g., no unconstrained cysteine unless intended, removing NXT glycosylation sites); and as mentioned, even doing in vitro selection for stability (like heat shock + protease treatment to filter out unstable clones). Some libraries are screened in their construction phase by expressing a sample in mammalian cells to ensure the clones can be produced a proactive QA step. The trend is toward libraries that yield antibodies with “drug-like” properties straight out of panning, reducing the need for extensive downstream engineering.
Advanced Display Platforms and Hybrids: We are also seeing innovation in how libraries are displayed and panned. For example, microfluidic selection systems have been introduced, where phage or yeast libraries are screened in picodroplets or on chips, allowing finer control and even coupling binding with functional assays (like signaling in a cell) in high throughput. There’s interest in cell-free selections that mimic some aspects of cell display e.g., ribosome display coupled with a downstream reporter assay. Another area is combining phage and yeast: one can do a broad phage selection, then transfer that pool to yeast display for a more granular sorting and affinity ranking (taking advantage of each platform’s strengths). Additionally, mammalian display methods have improved (viral delivery systems, display constructs, and automation) making it more feasible to sort 10^7 10^8 cells for binder discovery. In 2019, a study displayed a whole immune repertoire as IgG on mammalian cells and successfully isolated high-affinity antagonists by FACS, something that would have been extremely difficult five years prior.
Next-Gen Sequencing in all stages: While NGS in library work is not new, its use has become almost standard and even more sophisticated. In recent years, protocols for paired heavy-light sequencing have matured one can capture which heavy and light chain came from the same cell or phage, either by physical linkage or by computational grouping, enabling true repertoire reconstruction of Fab libraries by NGS. This has helped in analyzing natural libraries and ensuring that chain shuffling hasn’t created unnatural pairs that never actually pair in outputs (if they never appear together in any binder after selection, perhaps they were incompatible). Also, long-read sequencing (PacBio, Nanopore) is being applied to libraries to sequence full-length antibody inserts in one read. This avoids assembly errors and can handle very long CDR3s or full IgG libraries that short reads struggled with. The data from NGS is increasingly used for real-time decision making in campaigns some groups do quick turnaround NGS (within 24 hours) after a selection round to decide how to proceed for the next round (e.g., adjust antigen concentration or competitor based on which clones are enriching).
“Library of Libraries” approaches: A recent innovation in practice is to create multiple sub-libraries, each exploring a concept, and then either combine them or screen them separately. For instance, a team might build one library biased for long H3 loops, another for medium loops, another for short loops; or libraries each on a different framework (one on a VH3-23, one on a VH1-69, etc.). These sub-libraries can be panned in parallel. This way, instead of one giant pool, you have several pools each with an internal theme improving coverage of different structural categories. Twist Bioscience has discussed such strategies, where their massive DNA synthesis capacity is used to produce several distinct synthetic libraries (termed a “Library of Libraries”) to maximize the chance of finding an antibody for any given target. After selections, one can compare which sub-library yielded hits this gives insight into what design works best for that antigen (e.g., maybe long loops were needed for a deep pocket target, etc.). This approach also mitigates risk: if one library had an unforeseen flaw, the others still provide chances.
Nanobody and Non-traditional Scaffolds: While this review focuses on antibodies, another trend is the extension of library methods to other scaffolds (like V_HH camelid single domains, or even non-Ig proteins). Phage and yeast display of nanobody libraries have become very popular, including fully synthetic nanobody libraries designed with similar principles (biasing certain CDRs, etc.). Nanobodies have different CDR composition (only three CDRs, heavy-chain only) and can have extremely long CDR-H3 loops. New libraries have taken advantage of that by allowing 20 24 amino acid H3 loops with cysteine bonds, etc., to yield paratopes that can reach concave epitopes (e.g., enzyme active sites). The technical aspects remain analogous, but with some differences in cloning (often nanobody libraries are done as single domains so even larger diversities can be reached since only one gene instead of two in Fab). The success of a synthetic nanobody library developed in recent years (e.g., a 2022 report of a synthetic nanobody library yielding binders to multiple GPCRs) underlines that the design principles we discussed apply broadly.
Automation and Throughput: Finally, lab automation is influencing library workflows. Selection processes that used to be manual (like multiple rounds of panning) can be partially automated with liquid handlers and even robotics for colony picking, DNA prep, etc. High-throughput cloning techniques allow building multiple libraries or sub-libraries faster. Some groups are pursuing one-pot selection plus sequencing pipelines for instance, using emulsion setups where each bead gets antigen and a library member, then sorting beads and sequencing to get binder sequences in a single integrated process. Such techniques are still experimental but could shorten the cycle from library to hit.
Conclusion
The period 2020-2025 has been marked by a synthesis of rational library design, rigorous validation, and computational assistance, yielding libraries that are larger, cleaner, and more target-tailored than ever before. Trends like machine learning-guided design and developability filtering at the library stage show how the field is addressing past pain points (e.g., handling enormous sequence spaces and avoiding poor-quality hits). With these innovations, antibody libraries continue to be a cornerstone of biotechnological discovery, marrying diversity generation with precision engineering. As we move forward, one can envision truly smart libraries perhaps even ones that evolve in real-time with algorithmic guidance but even in their current state, the technical prowess behind constructing and harnessing antibody libraries is enabling the rapid discovery of binders for research and therapeutic use at an unprecedented pace.
References and Sources
1. McLaughlin L. Protein Libraries with Controlled Amino Acid Diversity, TRIM Technology. (Jan 14, 2025) LinkedIn | Biotechnology Reviews - https://www.biotechnologyreviews.com/p/protein-libraries-with-controlled
2. McLaughlin L. Designing TRIM Oligonucleotide Libraries: The Foundation of Precision Antibody Engineering (A-to-Z). LinkedIn | Biotechnology Reviews - https://www.biotechnologyreviews.com/p/antibodyprotein-libraries-with-trim
3. McLaughlin L. Antibody & Protein Library Codon Design: Avoiding PTMs and Restriction Sites. LinkedIn | Biotechnology Reviews - https://www.biotechnologyreviews.com/p/protein-and-antibody-engineering
4. McLaughlin L. Antibody Classifications, Antibody Discovery and Therapeutics. LinkedIn | Biotechnology Reviews - https://www.biotechnologyreviews.com/p/antibody-scaffolds
5. McLaughlin L. Exploring Trimer Phosphoramidite Technology for Antibody Library Generation. LinkedIn | Biotechnology Reviews - https://www.biotechnologyreviews.com/p/trim-technology-for-generating-protein
6. McLaughlin L. Generating Diversified ssDNA Libraries of Oligonucleotides for Antibody, Peptide and Nanobody Discovery and Therapeutics. LinkedIn | Biotechnology Reviews - https://www.biotechnologyreviews.com/p/generating-diversified-ssdna-libraries
7. Aban, A. et al. (2018). Rational library design by functional CDR resampling. (PNAS) Describes constructing a synthetic scFv library by recombining predefined CDR sequences from an antibody database, leading to improved hit ratespmc.ncbi.nlm.nih.govpmc.ncbi.nlm.nih.gov.
8. Ofran, Y. et al. (2016). Understanding differences between synthetic and natural antibodies. (mAbs 8:278-287) Observes that synthetic antibodies often rely predominantly on CDR-H3, which can limit epitope diversity (more antigen contacts confined to one CDR)researchgate.net.
9. Zhang, Y. (2023). Evolution of phage display libraries for therapeutic antibody discovery. (mAbs 15:2213793) Reviews three generations of phage libraries: from first-gen natural/naïve libraries to second-gen synthetic (with human germline frameworks, natural amino acid distributions) to third-gen with developability improvementspmc.ncbi.nlm.nih.govpmc.ncbi.nlm.nih.gov. Also discusses use of trinucleotide synthesis and combining natural CDR-H3 with synthetic diversitypmc.ncbi.nlm.nih.govpmc.ncbi.nlm.nih.gov.
10. Shim, H. (2015). Synthetic approach to the generation of antibody diversity. (BMB Reports 48:489-494) A review of synthetic library design strategies, diversification methods (degenerate oligos vs. defined mixtures), and comparisons of various libraries (HuCAL, etc.). Emphasizes balancing diversity with preserving antibody structure.
11. Matsunaga, R. & Tsumoto, K. (2025). Accelerating antibody discovery and optimization with high-throughput experimentation and machine learning. (J. Biomedical Science 32:46) A review highlighting the integration of large-scale data (NGS, high-throughput assays) with ML for antibody engineeringjbiomedsci.biomedcentral.comjbiomedsci.biomedcentral.com. Fig.1 from this paper illustrates the ML-guided antibody design pipeline used in this reviewjbiomedsci.biomedcentral.com.
12. Li, L. et al. (2023). Machine learning optimization of candidate antibody yields highly diverse sub-nanomolar affinity antibody libraries. (Nat. Commun. 14:3454) Demonstrates an ML-driven approach to affinity maturation: a language model + Bayesian optimization designed scFv libraries with 99% of variants improved over the parent and explored broader sequence space than conventional methodsnature.comnature.com.
13. Huang, C-Y. et al. (2023). Highly reliable GIGA-sized synthetic human therapeutic antibody library construction. (Front. Immunol. 14:1089395) Describes construction of a 2.5×10^10 scFv library (DSyn-1) on a single human scaffold with six CDRs diversified, using β-lactamase selection for each CDR to maintain functionalityfrontiersin.orgfrontiersin.org. Yields sub-nM TIM-3 binders, illustrating state-of-the-art library size and qualityfrontiersin.orgfrontiersin.org.
14. Mccafferty, J. et al. (1990). Phage antibodies: filamentous phage displaying antibody variable domains. (Nature 348:552-554) Classic paper that launched phage display of antibodies, showing a library of ~10^7 could isolate binding Fab fragments. Historical significance as proof of concept for in vitro antibody libraries.
15. Boder, E.T. & Wittrup, K.D. (1997). Yeast surface display for screening combinatorial polypeptide libraries. (Nat. Biotechnol. 15:553-557) Established yeast display technology, displaying scFv on yeast and isolating high-affinity mutants by flow cytometry. Demonstrated affinities can be improved by iterative sorting.
16. Fellouse, F.A. et al. (2007). Mixing and matching of V(H) domains and V(L) domains for the design of an antibody library. (Proc. Natl. Acad. Sci. USA 104: 8456-8461) An example of semisynthetic library construction by combinatorial pairing of heavy chains and light chains from different sources, illustrating the concept of gaining diversity through new pairings.
17. Sidhu, S.S. & Fellouse, F.A. (2006). Synthetic therapeutic antibodies. (Nat. Chem. Biol. 2:682-688) A review focusing on synthetic antibody libraries and how phage display can be used to derive therapeutic leads, discussing strategies like restricted diversity and consensus frameworks.
18. Ravn, U. et al. (2010). By-passing in vivo selection: building and screening combinatorial antibody libraries independent of immunization. (Biotechnology Journal 5: 14-21) Describes methods for building large naïve human libraries and panning them to isolate antibodies without immunization, touching on library quality and functional content.
19. Luginbühl, B. et al. (2020). De novo discovery of functional antibodies by next-generation sequencing of B cell repertoires. (Curr. Opin. Immunol. 65:21-27) Explores how NGS of immune repertoires and even machine learning on those repertoires is enabling in silico generation of antibody libraries or direct candidate discovery, reflecting the trend of computationally mining natural diversity.