

Natural Killer Cells: Biology, Function, and Modern Therapeutic Applications
Natural Killer (NK) cells are cytotoxic lymphocytes of the innate immune system with a remarkable capacity to recognize and eliminate virally infected and malignant cells without prior sensitization.

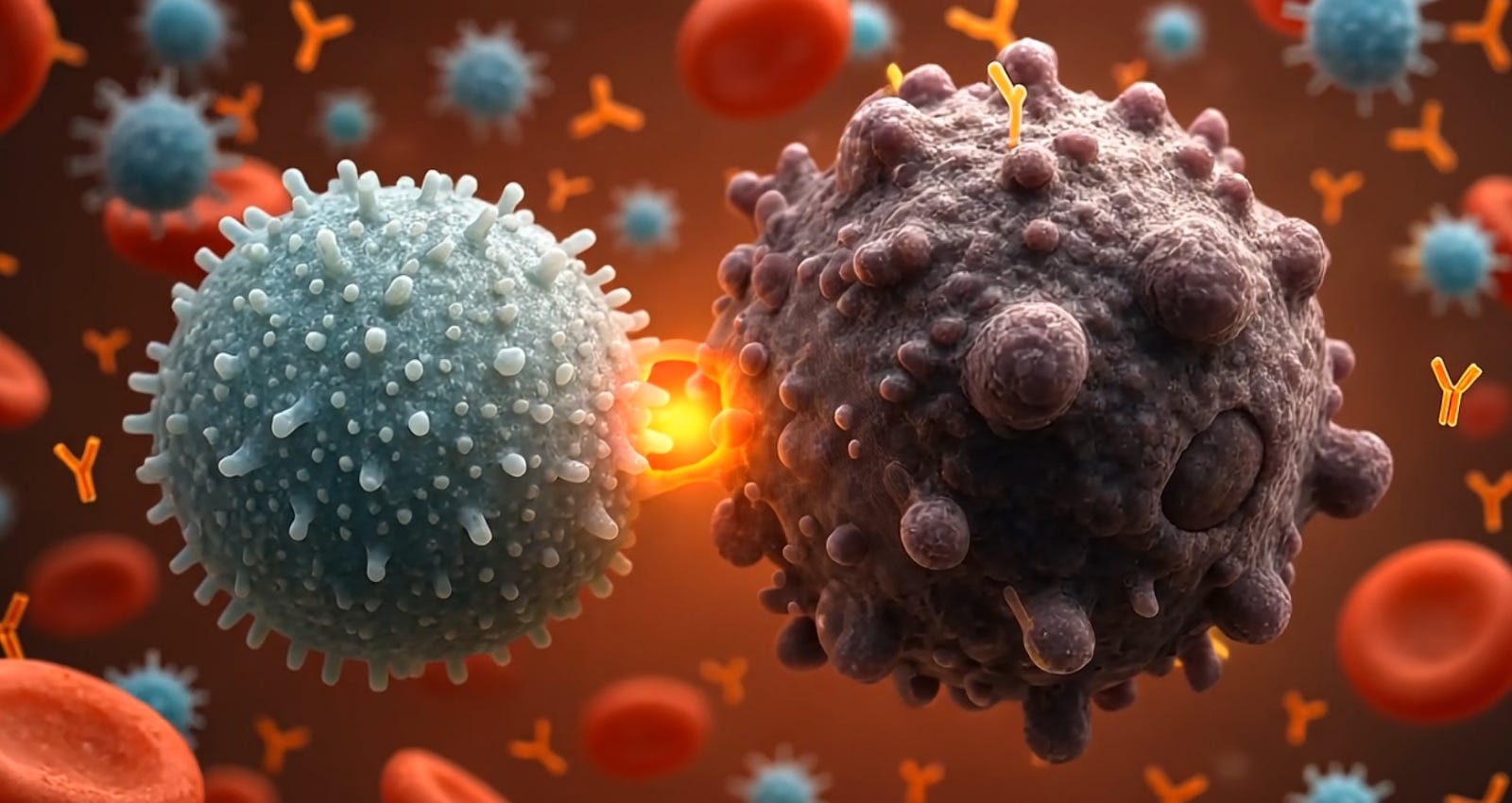
Natural Killer (NK) cells are cytotoxic lymphocytes of the innate immune system with a remarkable capacity to recognize and eliminate virally infected and malignant cells without prior sensitization. They comprise ~5–15% of circulating lymphocytes and serve as first-line effectors in immune surveillance against tumors and infections. Unlike T cells and B cells, NK cells do not rely on rearranged antigen-specific receptors; instead, they use a sophisticated array of germline-encoded activating and inhibitory receptors to detect cellular stress and “missing-self” signals on potential target cells. A delicate balance of these signals governs NK cell activation. When an NK cell becomes activated upon encountering an abnormal cell, it can directly lyse the target through cytotoxic mechanisms or indirectly modulate the immune response via cytokine secretion and crosstalk with other immune cells. These unique features, along with an inherent lack of requirement for prior antigen exposure, make NK cells attractive for therapeutic applications in cancer and other diseases.
In recent years, there has been surging interest in harnessing NK cells for adoptive cell therapy and engineering them with chimeric antigen receptors (CARs) to create “off-the-shelf” cancer treatments. Early clinical studies demonstrated that NK cell infusions can be safe and produce anti-tumor responses in patients with leukemia. Compared to T cell-based therapies, NK cell therapies offer distinct advantages: they do not cause graft-versus-host disease (GvHD) even when using allogeneic donors, they carry a lower risk of cytokine release syndrome (CRS) and neurotoxic side effects, and they can be prepared as standardized, cryopreserved products from renewable cell sources. Nonetheless, there are challenges to overcome – NK cells can be difficult to expand to therapeutic numbers, may exhibit short in vivo persistence, and face immune suppressive tumor microenvironments.
This article provides a comprehensive review of NK cell biology and the latest advances in translating NK cells into therapies. We first discuss NK cell development, phenotypes, and cytotoxic mechanisms. We then examine NK cells’ modes of action in immune surveillance and tumor recognition. Next, we detail approaches for adoptive NK cell transfer, including sources, expansion methods, and genetic engineering strategies such as CAR-NK cells. We explore the emerging field of stem cell–derived NK cells as a platform for scalable “off-the-shelf” therapies. We also highlight NK cell culture techniques relevant to clinical manufacturing. Finally, we review combination strategies (with antibodies, cytokines, or checkpoint inhibitors) and survey the current landscape of clinical trials using NK cells against cancer. Throughout, we cite key findings from immunology research and clinical studies to provide an expert-level understanding of how NK cells are being leveraged in biotechnology and medicine.
NK Cell Biology: Development, Subsets, and Receptors
Origin and Development: NK cells originate from the common lymphoid progenitor in the bone marrow and develop along a lineage distinct from T and B cells. Interleukin-15 (IL-15) is the critical cytokine driving NK cell development and maturation, as it promotes survival and proliferation of NK progenitors (CD122^+ NK precursors) in the bone marrow. Immature NK cells expressing markers like NKp46 undergo further differentiation and egress into circulation and tissues. Several transcription factors regulate NK development, including Eomes, T-bet (TBX21), and ID2, which coordinate the acquisition of NK cell effector functionsf. Uniquely, NK cells do not undergo V(D)J recombination and thus do not have clonotypic antigen receptors. Instead, they express an array of germline-encoded receptors that are either activating or inhibitory. NK cell “education” (or licensing) is the process by which interactions of inhibitory NK receptors with self MHC class I molecules tune the NK cell’s responsiveness, ensuring tolerance to healthy self while retaining reactivity to “missing-self” targets (cells that lack self MHC)j.
CD56^bright vs CD56^dim Subsets: In humans, NK cells are broadly classified into two major subsets based on CD56 surface density. CD56^bright NK cells constitute a smaller fraction (~5–10%) of blood NK cells and are abundant in lymphoid tissues; they are less mature, lack or weakly express the Fc receptor CD16, and are characterized by potent cytokine production (e.g. IFN-γ, TNF-α) rather than cytotoxicity. CD56^dim NK cells, in contrast, make up the majority (~90%) of circulating NK cells and represent a more mature subset; CD56^dim NKs strongly express CD16 and are highly cytotoxic, capable of natural killing and antibody-dependent cell-mediated cytotoxicity (ADCC). Evidence suggests CD56^bright cells can differentiate into CD56^dim cells under the influence of cytokines like IL-15 and IL-2. CD56^dim NK cells also upregulate markers of terminal differentiation such as CD57 and KIRs (killer immunoglobulin-like receptors). Besides these two subsets, tissue-resident NK cells exist in organs (e.g. liver, uterus) with distinct phenotypes (such as CD49a^+ in the liver, or decidual NK cells in the uterus) and specialized roles. Overall, the CD56^bright subset is viewed as immunoregulatory and prolifically secretes cytokines, whereas CD56^dim NKs are the principal cytotoxic effectors in peripheral blood.
Activating and Inhibitory Receptors: NK cell function is dictated by a balance between activating signals (which trigger killing) and inhibitory signals (which restrain NK activity to prevent damage to healthy cells). NK cells express numerous activating receptors that recognize stress-induced ligands on target cells. These include the natural cytotoxicity receptors NCRs (NKp30, NKp44, NKp46), NKG2D, and co-activating receptors like DNAM-1 (CD226), 2B4, and SLAM-family receptors. For example, NKG2D on NK cells binds to induced self ligands such as MICA/B and ULBP proteins, which are upregulated on infected or transformed cells. Ligation of activating receptors initiates signaling cascades involving ITAM-bearing adaptor molecules (e.g. CD3ζ, DAP12) and tyrosine kinases (ZAP70, SYK), ultimately leading to exocytosis of lytic granules and target cell apoptosis. NK cells also express CD16 (FcγRIIIa), especially on CD56^dim cells, which binds to the Fc portion of IgG antibodies on opsonized targets and triggers ADCC – a major mechanism by which therapeutic antibodies enlist NK cells to kill tumor cells Counterbalancing these are inhibitory receptors that survey for normal self markers. Chief among them are the KIRs (killer immunoglobulin-like receptors) and the heterodimer CD94/NKG2A, which recognize MHC class I molecules on potential target cells Each individual has a unique repertoire of KIR genes (there are 14 KIR genes in humans) encoding receptors that typically have long cytoplasmic tails transmitting inhibitory signals upon binding “self” HLA class I ligands. If an NK cell’s inhibitory receptors engage sufficient MHC I on a cell, they deliver dominant negative signals that prevent killing – this is how healthy cells are spared. However, virus-infected or malignantly transformed cells often have downregulated MHC I (the phenomenon of “missing-self”), making them prone to NK cell attack due to the lack of inhibitory signalingj. In addition, cellular stress or oncogenic transformation can upregulate ligands for activating receptors (like NKG2D ligands), tipping the balance toward NK cell activation. Through this array of receptors, NK cells continually scan host tissues for any imbalance in “self” vs “stress” signals and rapidly eliminate cells that are deemed a threat.
Cytotoxic Mechanisms: Once activated, NK cells employ multiple cytotoxic strategies to kill abnormal cells. A primary mechanism is the release of lytic granules containing perforin and granzymes. Upon formation of an immunological synapse with a target cell, the NK cell’s microtubule-organizing center and granules polarize toward the target, and perforin is released to form pores in the target membrane. Through these pores, granzymes (serine proteases) enter the target cell cytosol and induce apoptosis by cleaving caspases and other substrates. This granule exocytosis pathway can induce rapid cell death within hours and is analogous to cytotoxic T lymphocyte killing. NK cells also express death ligand receptors that trigger apoptosis in target cells upon engagement. Notably, NKs can upregulate Fas ligand (FasL) and constitutively express TRAIL (TNF-related apoptosis-inducing ligand); binding of FasL to Fas (CD95) or TRAIL to DR4/DR5 on target cells activates the extrinsic death receptor pathway of apoptosis. Importantly, only FasL and TRAIL have been shown to act as direct cytotoxic effector molecules for NK-mediated killing in humans and mice. In some contexts, NK cells preferentially use the death-receptor pathway (which is a somewhat slower kill mechanism) after sequential encounters with targets, possibly as a means to conserve granules during serial killing. Additionally, through ADCC, NK cells recognize antibody-coated targets via CD16 and release granules to kill the opsonized cell. Aside from direct cytolysis, NK cells secrete inflammatory cytokines (notably IFN-γ and TNF-α) upon activation. IFN-γ released by NK cells can have potent anti-tumor and anti-viral effects: it directly inhibits proliferation of some tumor cells, induces upregulation of MHC class I on surrounding cells, and activates macrophages and Th1 adaptive responses. NK-derived cytokines and chemokines also help shape subsequent immune responses – for example, recruiting dendritic cells and T cells to sites of infection or tumor. In summary, NK cells are equipped with a versatile “armamentarium” of cytotoxic molecules and immunomodulators. They can eliminate target cells by granule-mediated apoptosis or death receptor pathways and can orchestrate broader immunity via cytokine secretion. This cytotoxic versatility underlies their critical role in immunosurveillance and is being exploited in therapeutic contexts.
Mechanisms of NK Cell Action in Immune Surveillance
NK cells function as vigilant sentinels that patrol the body for cells undergoing stress, transformation, or infection. Immune Surveillance and Missing-Self Recognition: Kärre’s “missing-self” hypothesis, proposed in the 1980s, elegantly explained how NK cells detect cells that lack normal expression of self MHC class I. Healthy cells express MHC I molecules which engage NK cell inhibitory receptors (KIRs, NKG2A), signaling the NK cell to refrain from killing. Cells that lose MHC I – a common evasion strategy of viruses and tumors – fail to trigger those inhibitory signals, thereby allowing NK cell activation and cytotoxicity. Concurrently, stressed or transformed cells often upregulate ligands for NK activating receptors, such as MICA/B and ULBP1-6 for NKG2D, or nectin/Poliovirus receptors for DNAM-1, which serve as “altered-self” flags that NK cells recognize. Through the integration of missing-self (lack of inhibition) and altered-self (presence of activation) signals, NK cells achieve a form of discriminative surveillance, eliminating cells that are either invisible to T cells (due to absent MHC I) or overtly abnormal. This places NK cells at the forefront of defense against certain viruses (e.g. cytomegalovirus) and against tumors that downregulate MHC I to evade T cells. Indeed, studies in both mice and humans have shown that individuals with NK cells lacking inhibitory KIRs for the host’s MHC (i.e. KIR/KIR-ligand mismatch) have stronger NK anti-leukemia reactivity, supporting the importance of missing-self recognition in clinical outcomes.
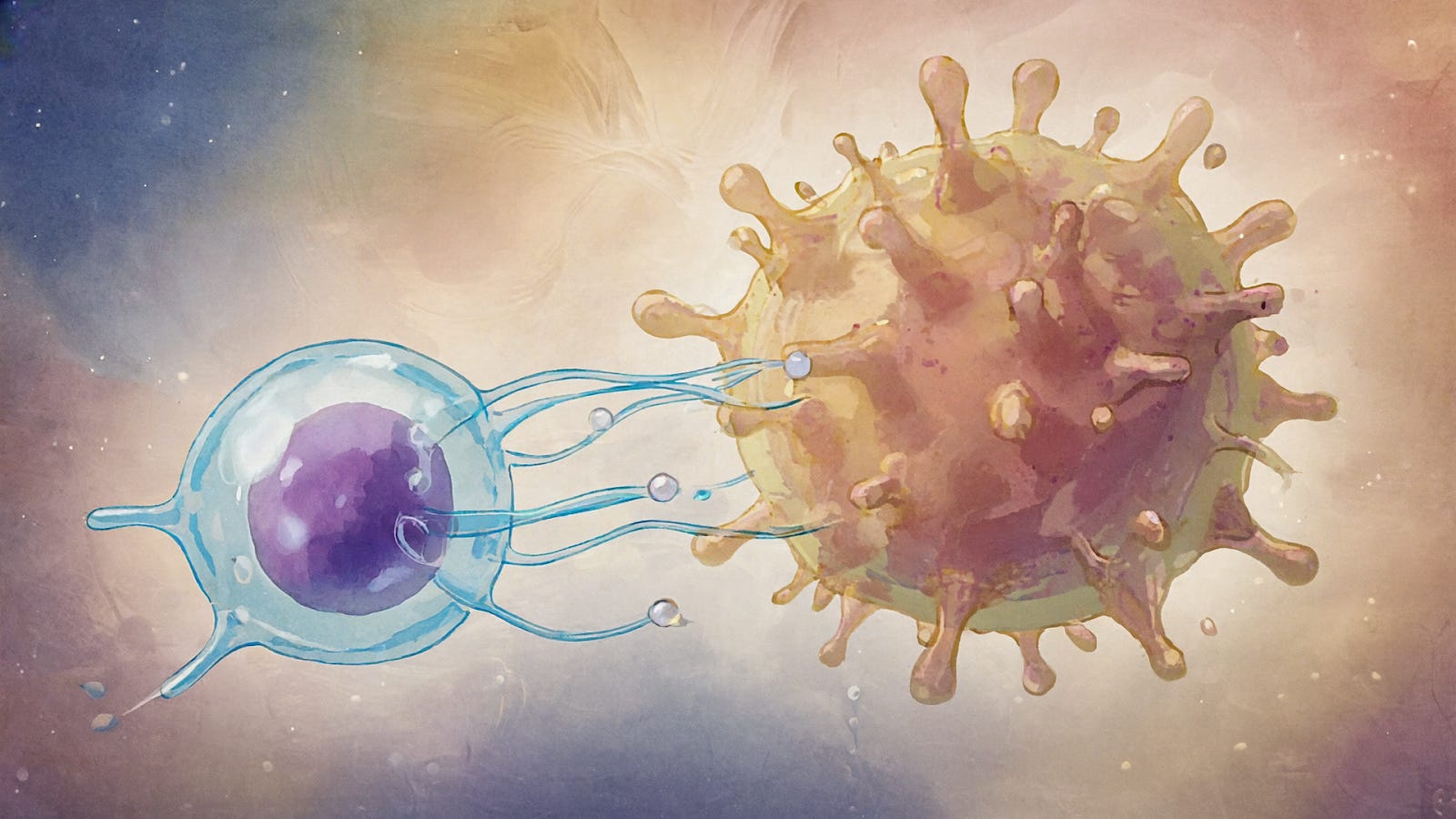
Immunological Synapse Formation: When an NK cell recognizes a susceptible target cell, it forms an immune synapse – a specialized contact interface – to focus its attack. Receptors on the NK cell bind ligands on the target, and adhesion molecules (like LFA-1 interacting with ICAMs) secure the tight conjugation. Inside the NK cell, signaling leads to actin cytoskeleton reorganization and polarization of lytic granules towards the synapse. The contents of granules (perforin and granzymes) are then secreted in a confined space at the synaptic cleft, concentrating the lethal hit on the target cell while sparing bystander cells. The integrity of this lytic synapse is crucial – for instance, if tumor cells induce an altered (e.g. protease-cleaved) form of ligands or modulate the NK cell’s ability to form a mature synapse, they can resist NK killing. NK cells can form different types of synapses: an activating lytic synapse with a susceptible target, or an inhibitory synapse (often characterized by different molecular organization) when encountering a healthy cell with normal MHC I. Quantitative imaging studies have revealed that NK cells integrate signals over the contact area and make a rapid “decision” to kill within minutes of synapse formation. The efficiency of lytic synapse formation is one factor that determines NK cell cytotoxicity and is influenced by both NK cell intrinsic factors (e.g. actin dynamics, signaling thresholds) and target cell properties (e.g. surface ligand density, cell stiffness). Importantly, after delivering a lethal hit, NK cells can detach and recycle to engage other targets – a single NK cell is capable of serial killing, though with some decline in cytotoxic speed as granule stores are used and replenished.
Recognition of Infected and Transformed Cells: NK cells play a pivotal role in controlling infections, especially by certain viruses and intracellular bacteria, as well as early in tumor development. Virally infected cells often present a paradox to the immune system: while cytotoxic T cells look for viral peptides on MHC I (which some viruses suppress), NK cells are activated by the absence of MHC I and by stress-induced ligands upregulated during infection. For example, CMV encodes proteins that downregulate HLA class I to evade T cells, but this renders the infected cell susceptible to NK killing (“missing-self”). In addition, interferons and other inflammatory signals during infection induce increased expression of activating ligands (like ULBP family) on infected cells, flagging them for NK cells. NK cells also express receptors for conserved viral molecules; for instance, NKp46 can recognize hemagglutinin on influenza-infected cells. In the context of tumors, cells undergoing malignant transformation often experience DNA damage or oncogenic stress responses that result in expression of NKG2D ligands (MICA, MICB, etc.) and other danger signals on the tumor cell surface. Early-stage tumors or metastases can thereby be recognized and eliminated by NK cells before an adaptive immune response is mounted – a process termed cancer immunosurveillance. Epidemiologic studies have correlated higher NK cell activity with lower cancer risk, and murine models lacking NK cells develop tumors more readily. NK cells are particularly critical in preventing the outgrowth of tumors that have lost MHC class I (escaped T cell immunity) or that exist in contexts where T cells are not yet primed (e.g. “cold” tumors). Furthermore, NK cells contribute to immune editing of tumors: by killing highly immunogenic tumor cells, they may select for tumor variants that become more stealthy over time.
Cytokine Secretion and Immune Modulation: Beyond direct cytotoxicity, NK cells secrete a range of cytokines and chemokines upon activation, which shape the subsequent immune response. Prominent among these is IFN-γ, which NK cells produce rapidly (often within 2–4 hours of stimulation). IFN-γ from NK cells activates macrophages to enhance phagocytosis and cytokine production, and it helps drive Th1 polarization of CD4 T cells and boosts CD8^+ T cell responses. It also has direct anti-proliferative and apoptotic effects on certain tumor cells and can increase their expression of MHC I, paradoxically making tumors more visible to T cells but also potentially more resistant to NK cells (since high MHC I engages NK inhibitory receptors). NK cells also secrete TNF-α, which can induce apoptosis in some target cells and activate endothelial cells to recruit immune cells. Additionally, NK cells can produce growth factors (GM-CSF) and a variety of chemokines (CCL3, CCL4, XCL1, etc.) that attract dendritic cells, T cells, and other immune cells to the site of attack. Interestingly, NK cells have been found to sometimes secrete immunosuppressive cytokines as well – for example, a subset of resting NK cells can secrete TGF-β and IL-10, especially in certain contexts like early pregnancy or cancer, which may dampen inflammation. These immunoregulatory outputs might serve to prevent excessive tissue damage or autoimmunity. In tumors, however, such NK-derived TGF-β and IL-10 can contribute to an immunosuppressive milieu. Overall, through cytokine and chemokine release, NK cells orchestrate a multi-cellular response: they enhance antigen presentation (by recruiting and maturing dendritic cells), promote adaptive immunity, and also directly influence other innate cells (e.g. activating macrophages, influencing neutrophils). Thus, NK cells act both as killers and as helpers in the immune system, bridging innate and adaptive immunity. This dual functionality is being harnessed in therapies – for instance, using cytokine pre-activated NK cells (e.g. IL-12/15/18-induced memory-like NK cells) to achieve heightened and sustained responses in cancer patients.
In summary, NK cells contribute to immune defense through: (1) Constant immune surveillance, detecting cells with missing-self or induced stress ligands; (2) Formation of immunological synapses to deliver targeted lethal hits via perforin/granzyme and death ligand pathways; (3) Rapid cytokine-mediated modulation of the environment, recruiting and activating other immune effectors. These mechanisms underscore why NK cells are an appealing tool in immunotherapy – they can seek and destroy abnormal cells broadly (not restricted by a single antigen), and they can amplify overall anti-tumor/anti-viral immunity. Harnessing these properties for therapeutic benefit requires overcoming some natural limitations of NK cells (such as their short lifespan and potential inhibition by the tumor microenvironment), which has prompted the development of strategies for NK cell expansion, activation, and genetic modification as described below.
Adoptive NK Cell Transfer: Isolation, Expansion, and Cell Sources
Adoptive transfer of NK cells involves collecting NK lymphocytes from a donor source, expanding/activating them ex vivo, and infusing them into a patient as a cellular therapy. Early clinical trials of NK cell infusions (dating back nearly two decades) established the feasibility and safety of this approach, with evidence of anti-tumor activity in leukemia patients. However, manufacturing a therapeutic NK cell product presents unique challenges: NK cells constitute only a small fraction of peripheral blood lymphocytes (~5–15%), they do not proliferate as readily as T cells in vivo, and they can be inhibited by host factors if not properly prepared. Below we outline the methods for NK cell isolation and expansion, and the various sources of NK cells used in therapy.
Isolation of NK Cells: The most common source for clinical NK cell therapy has been peripheral blood from donors or patients. Large-volume leukapheresis is performed to collect mononuclear cells, from which NK cells (CD56^+CD3^- fraction) must be enriched. Enrichment is critical because contaminating T cells could cause GvHD (if from an allogeneic donor) or expand undesirably, and B cells or tumor cells in the product could pose risksj. Two main isolation techniques are employed: (1) Immunomagnetic selection using clinical-grade systems like CliniMACS®. This can be done either by positive selection for CD56^+ cells or by negative depletion of CD3^+ T cells (and CD19^+ B cells). Often a combination is used – first deplete T cells and B cells, then enrich NK cells – to achieve a highly pure NK cell product. For example, a CD3/CD19 depletion followed by CD56 enrichment can yield a product that is >90% NK cells. Interestingly, some protocols report that simply depleting T cells (negative selection) yields better NK expansion than positive isolation, possibly because residual accessory cells (monocytes) in the product provide growth support in culture. (2) Density gradient or rosette-based separation (e.g. using RosetteSep™) can also enrich NK cells by aggregating undesired cells with antibody complexes and removing them during Ficoll gradient separation. While effective, these are often research-use methods; CliniMACS remains a workhorse for GMP-compliant cell separations. After enrichment, the NK cell product is typically >50–70% pure NK cells (CD56^+CD3^-), and further culture can purify it to >90% NK as T cells do not expand well under NK-friendly conditions. Alternative starting sources like umbilical cord blood units (which contain 5–20% NK cells naturally) can also be processed by immunomagnetic isolation of CD56^+ cells or CD34^+ progenitors (see below). Regardless of source, the isolated NK cells are generally in a resting state and require ex vivo activation and expansion to generate sufficient numbers for therapy (typical therapeutic dose range is 5×10^6 to 1×10^8 NK cells per kg of body weight).
Ex Vivo Expansion and Activation: Culturing NK cells with the appropriate stimuli can massively expand their numbers and boost their cytotoxic activity. NK cells by themselves have limited proliferation, so expansion protocols rely on feeders, cytokines, or artificial stimulants to drive NK cell growth over 1–3 weeks. A widely used approach is co-culture with irradiated feeder cells that express membrane-bound stimulatory molecules. The most common feeder is the K562 cell line (a human leukemia line) engineered to express membrane-bound IL-15 (or IL-21) and co-stimulatory ligands like 4-1BBL (CD137L). Dario Campana and colleagues pioneered a K562-based artificial antigen-presenting cell (aAPC) expressing 4-1BBL and mbIL-15 (designated K562-mbIL15-41BBL), which was shown to induce robust NK expansion (often >100-fold) and enhance cytotoxicity. Variants of this system (K562 cells with mbIL-21 instead of IL-15, or additional co-stimulatory molecules like CD86) have been adopted by multiple groups and companies to manufacture NK cells. For example, a feeder cell termed “FC21” (K562 expressing mbIL-21 and 4-1BBL) was developed to support high expansion of NK cells in ~2 weeks. Feeder cell-based methods can produce tens of billions of NK cells from a single donor apheresis, although they introduce irradiated allogeneic cells into the culture that must be removed or will be cleared after infusion. Some newer protocols avoid cell-based feeders by using feeder-free systems: these include plates coated with ligands (e.g. antibodies against NK cell activating receptors like NKp46/NKp30 and 4-1BB, along with Fc to engage CD16), or cell-derived nanoparticles that present membrane-bound IL-21. Feeder-free expansion typically requires a cocktail of cytokines to support NK growth. IL-2 was historically used at high doses to activate NK cells (as IL-2 can bind NK’s IL-2Rβγ receptors), but IL-15 is now favored for its more specific action on NK cells (and CD8 memory T cells) without stimulating Tregs. Many expansion protocols use IL-15 (e.g. 5–50 ng/mL) continuously, or IL-2 at high doses (1000–5000 IU/mL) every few days, often supplemented with IL-21 or IL-18 to enhance proliferation and cytotoxicity. A common cocktail for “cytokine-only” expansion is IL-15 + IL-21, which can expand NK cells ~20- to 40-fold over 2–3 weeks with retained killing function. Moreover, adding co-stimulatory antibodies (such as anti-NKp46, anti-4-1BB) or toll-like receptor agonists to cultures can further boost NK expansion in feeder-free systems. Regardless of method, expanded NK cells typically exhibit an activated phenotype: increased expression of activating receptors (NKG2D, NKp44, etc.), upregulation of adhesion molecules, and enhanced production of perforin and granzyme. Notably, expansion with IL-15 drives the differentiation of some NK cells toward an effector memory-like state with high cytotoxicity. In some protocols (e.g. for memory-like NK cells), NKs are briefly primed with cytokines like IL-12, IL-15, IL-18 for 16 hours, then rested – upon reactivation these “CIML” NK cells demonstrate heightened responses and are being tested clinically for AML. In summary, ex vivo expansion is a crucial step that transforms a small starting NK population into a therapeutically relevant dose. It can be achieved with irradiated feeder cells presenting membrane-bound growth signals or with defined media supplemented by cytokines and stimulatory reagents. The choice of expansion method may impact the phenotype: for instance, IL-21-based feeders tend to sustain a less differentiated, highly proliferative NK population, whereas prolonged high-dose IL-2 can drive NK cells to an exhausted state if not carefully optimized. Manufacturers must also ensure that any feeder components are removed or safe (K562 feeders are often irradiated at >100 Gy and will not proliferate in the patient, but they could theoretically trigger anti-allo responses).
Sources of NK Cells: One advantage of NK cells for therapy is that, unlike T cells which are usually patient-specific (autologous) products, NK cells can be sourced allogeneically from healthy donors without causing GvHD. This allows for off-the-shelf NK cell products and the use of robust donors. Major sources being utilized include:
Peripheral Blood NK Cells (PB-NK): Mature NK cells from adult blood donors are widely used. They require leukapheresis and isolation as described above. PB-NK cells are readily available and have a predominance of CD56^dim cytotoxic NK subset. Most non-engineered NK cell therapies in clinical trials to date have used PB-derived NK cells. Autologous NK cells (from the patient) have been infused in early trials as well, but these can be less effective if the patient’s NK cells are dysfunctional or inhibited by self-MHC recognition. Therefore, many trials now use allogeneic PB-NK from related or unrelated donors. The typical yields are on the order of a few x10^9 NK cells from one leukapheresis after expansion, sufficient for multiple doses.
Umbilical Cord Blood NK Cells (CB-NK): Cord blood contains a higher frequency of NK cells (15–30% of lymphocytes) that are phenotypically more immature. UCB-derived NK cells often express high levels of NKG2A and low levels of KIRs and adhesion molecules, reflecting their neonatal origin. Cord blood units can be banked and used as starting material without donor risk. One limitation is that the absolute number of NK cells in a single cord unit is relatively low, so significant ex vivo expansion is required to reach dose. Nevertheless, cord blood NK cells have been used successfully in both non-engineered and CAR-NK cell trials. A landmark first-in-human CAR-NK trial at MD Anderson Cancer Center employed cord blood-derived NK cells expressing an anti-CD19 CAR, IL-15, and a safety switch, achieving high response rates in lymphoma/leukemia patients with no CRS or GvHD. Cord NK cells may have greater proliferative capacity and a different KIR repertoire that is often alloreactive (KIR mismatch with recipients is common), potentially enhancing anti-tumor activity.
NK Cell Lines (e.g. NK-92): There are several immortalized NK or NK-like cell lines, of which NK-92 is the most prominent in clinical development. NK-92 was derived from an NK cell lymphoma and can be expanded indefinitely in culture with IL-2 support. It has a homogenous phenotype (CD56^+ CD16^- CD3^- and lacks most inhibitory KIRs except NKG2A) and robust cytotoxicity against a range of targets. The advantages of NK-92 are its ease of expansion and genetic modification, and the ability to generate a uniform product. However, since it is a tumor-derived line, it must be irradiated prior to infusion to prevent engraftment or outgrowth of the cell line in patients. Irradiation (usually 10 Gy) limits NK-92’s lifespan to a day or two in vivo, so the cells can kill targets but cannot proliferate in the patient. Despite this, clinical trials have shown NK-92 infusions to be safe and occasionally clinically effective (e.g. some partial responses in melanoma and renal cell carcinoma were reported in early studies). Several companies are developing CAR-NK-92 variants (NK-92 cells engineered with CARs) targeting antigens like CD19, HER2, EGFR, etc., which can be mass-produced and cryopreserved. One such product (t-haNK, targeting PD-L1 and EGFR) is in trials for solid tumors. NK-92’s lack of CD16 means it cannot perform ADCC, but it produces ample perforin/granzyme and has the convenience of a standardized cell line. There are other cell lines (YT, NKL, KHYG-1, etc.) but NK-92 is so far the only one used clinically. The need for irradiation and resultant lack of persistence is a downside for durable efficacy, but it provides a safe transient killer cell product.
Cord Blood or Placenta-Derived Hematopoietic Stem Cells: Another approach is to take CD34^+ hematopoietic progenitor cells (HPCs) from cord blood or placental blood and differentiate them into NK cells ex vivo. By using defined cytokine cocktails (e.g. stem cell factor, Flt3L, IL-15, IL-7) and stromal feeder layers, researchers have generated functional NK cells from CD34^+ cells in ~3–5 weeks. The rationale is that starting from an upstream progenitor may avoid some of the replicative senescence seen in peripheral NK cells after extensive expansion. Indeed, one clinical product (Glycostem’s “oNKord”, derived from UCB CD34^+ cells) was tested in a Phase I trial for elderly AML and showed safety and possible anti-leukemia activity. Celularity Inc. has also developed a cryopreserved NK product (CYNK-001, aka taniraleucel) from placental CD34^+ cells, which has been tested in clinical trials for glioblastoma, AML, and COVID-19. These NK cells derived from HSCs resemble normal mature NK cells in phenotype and function. Large-scale manufacturing is feasible (some protocols report >1,000-fold expansion from CD34 to NK cells), and the advantage is the effectively unlimited starting material from cord blood banks. However, differentiation protocols are complex and lengthy, and any residual undifferentiated CD34^+ cells would need to be removed to avoid engraftment of non-NK lineages.
Induced Pluripotent Stem Cell-Derived NK Cells (iPSC-NK): A cutting-edge source is to use induced pluripotent stem cells as a starting point to derive NK cells. iPSC-derived NK cells can be considered in a class of their own (discussed in detail in the next section). Briefly, iPSC technology enables the creation of a renewable master cell bank from a single donor’s cells (e.g. a skin fibroblast reprogrammed to iPSC), which can then be differentiated into NK cells in unlimited quantities. This offers tremendous potential for standardization and scalability: all patients receiving the therapy could get the same iPSC-NK product from a clonal source with pre-defined attributes. Several biotech companies have active programs in iPSC-derived NK cells, with products now in Phase I trials (for example, Fate Therapeutics’ FT500, FT516, FT596, etc., which are iPSC-NK cells with various enhancements). iPSC-derived NK cells are typically homogeneous and can be genetically engineered at the iPSC stage to introduce CARs or other modifications. We will expand on this approach later, given its importance as a next-generation NK source.
Each source of NK cells has its pros and cons. Peripheral blood provides fully mature, highly cytotoxic NK cells but with donor-to-donor variability and a need for robust expansion. Cord blood offers an allogeneic, readily available source but with more naive NK cells requiring maturation. NK cell lines are easy to grow and standardize but cannot persist in vivo and must be irradiated. HPC-derived and iPSC-derived NKs allow creation of off-the-shelf products at scale, but involve complex manufacturing and safety considerations (e.g. ensuring no residual pluripotent cells in iPSC products). Despite these differences, all sources have shown the ability to mediate anti-tumor effects in preclinical models. Indeed, NK cells from healthy allogeneic donors (whether PB, CB, or iPSC-derived) are typically more functional than patient autologous NK cells, because cancer patients often have impaired NK activity or tumors that educate autologous NKs to be less reactive. Thus, many clinical efforts focus on allogeneic NK cell therapy, taking advantage of NK cells’ lack of GvHD to treat patients with cells from unrelated donors. Table summaries in recent reviews catalog dozens of NK products in trials, illustrating the breadth of sources and methods being pursued.
Activation Strategies: In preparing NK cells for therapy, besides numeric expansion, one also aims to achieve an optimal activation and differentiation state. Ex vivo expanded NK cells are often “activated” by exposure to IL-2 or IL-15 (and feeder cells) such that they express CD69, have released perforin/granzyme (and synthesized new granules), and display heightened cytotoxicity in vitro. Some protocols include a brief activation step prior to infusion – for instance, incubating NK cells overnight with IL-2 and a final addition of a biopharmaceutical like OKT3 (anti-CD3) to engage accessory cells, which was shown to increase NK cell cytotoxicity. However, continuous maximal activation can also lead to exhaustion, so there is a balance to be struck. A concept tested in trials is “arming” NK cells with antibodies or cytokines just before infusion. For example, incubating NK cells with a monoclonal antibody (that they will carry into the patient bound to their CD16) to facilitate ADCC immediately on infusion. Another approach is priming NK cells with IL-12, IL-15, IL-18 to create cytokine-induced memory-like NK cells (as mentioned), which have shown enhanced responses in leukemia patients.
In summary, the process of adoptive NK cell therapy involves: selecting the source of NK cells (autologous vs various allogeneic sources), isolating NK cells or progenitors, expanding and activating them ex vivo with feeders and/or cytokines to attain sufficient numbers and potency, and formulating the cell product for infusion. With these methods, NK cells on the order of 10^7–10^9 per dose can be manufactured, which are doses comparable to those used in CAR-T cell therapies. The next sections will discuss how genetic engineering is being layered on top of these adoptive NK platforms to enhance their targeting and activity.
Genetic Engineering of NK Cells: CAR-NK and Beyond
Genetic modification of NK cells is a burgeoning area aimed at endowing NK cells with improved tumor-targeting and persistence, analogous to the revolutionary CAR-T cell strategies. Advances in gene transfer techniques now enable introduction of chimeric antigen receptors (CARs) and other transgenes into NK cells, despite NK cells being historically difficult to transduce or transfect. The result is CAR-NK cells – NK cells that carry a synthetic receptor for specific antigens on tumor cells, triggering NK activation upon antigen binding. Engineering can also be used to knock out inhibitory genes or add supportive genes (like cytokines) to bolster NK cell function. Below we outline design principles for CAR-NK cells, gene delivery methods, notable gene targets, and how CAR-NKs compare to CAR-Ts.
CAR Design for NK Cells: A CAR construct typically consists of an extracellular antigen-binding domain (an scFv antibody fragment), a transmembrane hinge region, and intracellular signaling domains. First- and second-generation CARs used in T cells (with CD3ζ alone or CD28/4-1BB costimulatory domains) have been shown to also activate NK cells when expressed, because NK cells do possess some of the same signaling machinery (FcRγ and CD3ζ chains, etc.). Indeed, the clinical CD19-CAR used in the 2020 MD Anderson trial was a CD28-ζ CAR, and it effectively triggered NK cell cytotoxicity and cytokine release against CD19^+ leukemia. However, researchers are actively exploring CAR designs optimized for NK biology. NK cells have unique signaling adapters like DAP10 and DAP12 (used by NKG2D and natural cytotoxicity receptors) and co-activation molecules like 2B4 (SLAMF4) that signal via SAP family adaptors. One strategy has been to incorporate NK-specific signaling domains into CARs: for example, replacing CD3ζ with DAP12 or adding 2B4 (SLAMF7) endodomains. Preclinical tests showed that a CAR containing NKG2D’s transmembrane and DAP10 signaling domain plus CD3ζ could robustly activate NK cells. Similarly, CARs with a 2B4+ζ fusion signaling domain have shown superior NK activation compared to 4-1BB or CD28 in some systems. Another example is using DNAM-1 (CD226) intracellular domain as a co-stimulatory module in NK CARs, which was reported to outperform CD28-based CAR signaling in an NK-92 model targeting GPC3 on liver cancer. These findings highlight that the optimal signaling domain for CAR-NK might differ from CAR-T, and combinations of T cell signals (e.g. 4-1BB) with NK signals (e.g. 2B4, DAP10) are being screened for best functionality. Despite these innovations, many current CAR-NK trials still use second-generation CAR constructs originally developed for T cells, which appear to be quite effective in activating NK cells as well.
Another design consideration is the transmembrane domain of the CAR. CARs for T cells often use CD8α or CD28 transmembrane regions; in NK cells, using an NK cell receptor transmembrane (like NKG2D or NKp44) may improve stability and expression. For instance, an NKG2D-CAR (with NKG2D TM domain) was used in iPSC-derived NK cells and showed good surface expression and function.
Gene Delivery Techniques: Introducing genes into NK cells can be challenging because primary NK cells are relatively resistant to viral transduction and are non-dividing (except when stimulated). Nonetheless, several methods have proven successful:
Viral vectors: Retroviral and lentiviral vectors are widely used to stably transduce CAR genes into NK cellsf. Retroviruses (γ-retroviral vectors) require cell division for integration, so NKs need to be activated (IL-2-stimulated) and possibly briefly co-cultured with virus on retronectin-coated plates to achieve transduction. Lentiviral vectors (which can transduce non-dividing cells) have shown higher efficiency in transducing NK cells; typical protocols involve spinoculation of IL-15-activated NK cells with VSV-G pseudotyped lentivirus. Achieving 20–50% CAR expression in primary NK cells is considered good; NK cell lines (like NK-92) can reach >80% transduction. Adeno-associated virus (AAV) vectors have also been explored for NK cells, though less commonly, as they have smaller cargo capacity and tend to be used for in vivo gene delivery rather than ex vivo cell modification.
Electroporation and mRNA transfection: While viral integration yields permanent expression, some groups electroporate CAR-encoding mRNA into NK cells for transient expression (lasting a few days). This has the advantage of not requiring viral vectors and avoids genomic integration, but the short expression means repeated dosing would be needed. For example, cytokine-activated NK cells can be RNA-electroporated with CAR mRNA immediately prior to infusion – such CAR-NK cells can exhibit anti-tumor activity without the cost and time of viral vector manufacturing. This approach is being tested with products like “NKARTA’s NKX101” (an NKG2D CAR-NK) in which mRNA-electroporated donor NK cells are infused multiple times.
Nucleofection for gene knockout or knock-in: CRISPR/Cas9 technology has been successfully applied to NK cells by electroporating Cas9 ribonucleoproteins (Cas9 protein complexed with guide RNA) to knock out genes in resting NK cells. For instance, genes like CISH (a negative regulator of IL-15 signaling) have been knocked out to enhance NK cell responsiveness. This requires a highly optimized protocol to maintain viability, but studies have shown good knockout efficiency and subsequent expansion of the edited NK cells. More ambitiously, homology-directed repair to knock-in genes (e.g. inserting a CAR into a specific locus) in NK cells is under investigation, but the low proliferative capacity of primary NKs makes this challenging. iPSC-derived NK cells offer an easier platform for precise genetic modifications (done at the iPSC stage via CRISPR, followed by differentiation).
Transient proteins: Another non-viral method is using cell-penetrating peptides or nanoparticle delivery of DNA, but these are not common for NK yet.
Overall, lentiviral transduction remains the workhorse for manufacturing CAR-NK cells, with several clinical trials using lentivirus-modified NK or cord blood NK to express CARs. Retroviral methods are used for NK cell lines and sometimes primary NK (e.g. the GTMP facility protocols). The gene editing (CRISPR) approaches are mostly preclinical but hold promise for next-gen engineered NK products – for example, simultaneously inserting a CAR and knocking out multiple inhibitory receptors.
Enhancements and Targets in CAR-NK Cells: The genetic engineering of NK cells is not limited to CARs. A number of enhancement strategies are being pursued:
Cytokine support (“Armored” CAR-NKs): One limitation of NK cells is their short lifespan in vivo. To address this, CAR-NK cells have been engineered to secrete or express cytokines that support NK survival, most notably IL-15. Membrane-bound IL-15 (mbIL15) can be co-expressed with a CAR, providing autocrine and paracrine stimulation to the NK cells. The CAR19/IL-15 cord blood NK cells from MD Anderson included a secretory IL-15 linked via a 2A peptide in the CAR construct, enabling the CAR-NK cells to persist longer without exogenous cytokine support. Other groups have used mbIL21 co-expression in feeder-expanded NK cells to maintain their proliferation. Another strategy is to express dominant-negative TGF-β receptors or IL-18 receptors to make NK cells resistant to suppression and enhance their activation in tumors.
Suicide switches: To improve safety, genes like inducible caspase-9 (iC9) have been inserted so that the cells can be selectively destroyed if severe adverse events occur. The iC9 system, activated by a small molecule (AP1903), was included in the cord blood CAR-NK product and is being included in some allogeneic NK products to guard against unanticipated toxicity.
Target selection: CAR-NK cells are being designed against many of the same targets as CAR-T. Hematologic malignancies have been prime targets – e.g. CD19 for B-cell leukemia/lymphoma (where CAR-NK cells have shown complete remissions without GvHD or CRS), CD20 for lymphoma, CD33 and CD123 for acute myeloid leukemia, BCMA for multiple myeloma, etc. Solid tumor targets like GD2 (neuroblastoma), HER2 (breast/GBM), EGFR (glioblastoma), EpCAM, Mesothelin, PSMA, and others are also in development with CAR-NK cells. One interesting approach is a CAR that leverages an NK activating receptor as the targeting moiety: an example is a CAR that is actually a chimeric NKG2D receptor. In this design, the NK cell is engineered to overexpress a receptor (NKG2D) that recognizes multiple ligands on tumors; this was tested in NK cells (and T cells) to target the many tumors that express MIC-A/B or ULBPs. Another approach uses TriKEs and BiKEs (bispecific and trispecific NK cell engagers) to direct NK cells to targets without genetic engineering, but those are pharmacologic, not genetic, strategies.
Persistence and homing: NK cells typically have transient persistence after infusion (days to a couple of weeks). Genetic tricks to extend this include telomerase expression (to delay senescence) or downregulating inhibitory checkpoints (like knock-out of CIS, the CISH gene, which restrains IL-15 signaling – this knockout was shown to enhance NK cell metabolic fitness and anti-tumor function). Knocking out TIPE2 (a negative regulator of NK activation) was also found to unleash NK anti-tumor activity in combination with CISH knockout. To improve homing to solid tumors, chemokine receptors can be introduced – e.g. CXCR2 engineered NK cells showed better migration to renal carcinoma secreting CXCL2. CRISPR was used to delete NKG2A in NK cells to reduce inhibition by HLA-E and that improved tumor control in a mouse model. Similarly, knocking out adenosine A2A receptor (to prevent suppression by tumor-generated adenosine) and knocking out TGF-β receptor are strategies to make NK cells more resistant to common tumor microenvironment suppressive factors.
In essence, the genetic engineering toolbox for NK cells is expanding: CARs for targeting, cytokine genes for support, suicide genes for safety, checkpoint deletions for disinhibition, and chemokine receptors for homing are all being tested. Early clinical reports are encouraging – for example, in Phase I studies, CD19 CAR-NK cells induced responses in a majority of patients with lymphoma/leukemia, while exhibiting an excellent safety profile (no CRS or neurotoxicity). This contrasts with CAR-T cells, which, though effective, often cause severe CRS/ICANS.
Comparison with CAR-T Cells: CAR-NK cells inevitably invite comparison to CAR-T cell therapies. Both are forms of adoptive cell immunotherapy employing engineered lymphocytes, but NK cells have intrinsic differences:
Safety: CAR-NK cells have not been associated with significant CRS or ICANS in early trials. For instance, in a clinical study of cord blood CAR-NK cells in 11 patients, none developed CRS or neurotoxicity. This may be because NK cells secrete lower amounts of IL-1 and IL-6 (cytokines implicated in CRS) and because they lack clonal expansion kinetics of T cells. NK cells also undergo apoptosis if over-activated, potentially self-regulating their expansion. The absence of GvHD risk is a major advantage – NK cells do not attack healthy tissues based on allogeneic HLA differences, since their recognition is not MHC-restricted in the way TCRs are. This allows CAR-NK cells from unrelated donors to be given without HLA matching and without causing GvHD, a feat not possible with conventional T cells (unless TCR or HLA genes are deleted from CAR-T). Allogeneic CAR-T strategies must remove TCRs or use gene edits to prevent GvHD, whereas allogeneic CAR-NK can be used “as is.” Additionally, CAR-NK cells do not engraft long-term, which may reduce the risk of any delayed adverse events or insertional mutagenesis (in case of viral vectors). The flipside is that lack of long-term persistence might limit durable efficacy, but it greatly simplifies safety monitoring.
Efficacy and Persistence: CAR-T cells are known for their ability to proliferate and form long-lived memory cells in patients, sometimes persisting for years. NK cells typically have shorter persistence – weeks at most for primary NKs, and only days for irradiated NK cell lines. This means CAR-NK therapies might require multiple doses or adjunctive cytokine support (e.g. giving IL-2/IL-15 to patient) to sustain them. Early trials have shown impressive initial response rates with CAR-NK (e.g. rapid tumor reductions), but some responses have been transient, with relapses occurring once the CAR-NK cells disappear. Efforts to extend CAR-NK longevity include adding IL-15 to the CAR construct (as discussed) and repeat dosing schedules. Some Phase I trials are exploring giving CAR-NK infusions weekly or biweekly, which is feasible since these cells can be from an allogeneic batch (whereas CAR-T is usually a one-time autologous infusion). NK cells do not form true memory in the classical sense (except some evidence of “adaptive” NK cells in CMV infection), so the concept of long-term immunological memory is less applicable – although memory-like NK cells may persist a few months post-infusion in some studies.
Manufacturing: CAR-T therapies (except newer allogeneic CAR-Ts) are individually manufactured for each patient, a time-consuming and costly process. CAR-NK cells offer the possibility of an off-the-shelf inventory. A single donor or an iPSC line can yield dozens or hundreds of doses of CAR-NK cells that are cryopreserved and ready to use when a patient needs therapy. This dramatically reduces vein-to-vein time and could lower cost if production is scaled. NK cells can be banked and do not necessarily need to come fresh from the patient, which is particularly advantageous for patients who are lymphopenic or heavily pretreated. Moreover, CAR-NK manufacturing can be seamlessly integrated with existing bioreactors and expansion methods for NK cells.
Antitumor Mechanisms: CAR-T cells kill targets mainly via perforin/granzyme and FASL pathways as well, but NK cells bring additional mechanisms to the table. A CAR-NK cell not only kills through the CAR (which provides a strong activating signal) but can also simultaneously kill surrounding tumor cells through natural cytotoxicity and ADCC mechanisms that are independent of the CAR. For example, a CAR-NK targeting a tumor antigen might engage one tumor cell, and in the process, if that NK cell’s CD16 encounters therapeutic antibody opsonizing another tumor cell, it can kill that cell too (CAR-independent killing). This bystander killing could potentially address antigen heterogeneity in tumors – something CAR-T cells struggle with unless they are armored with cytokines to recruit other effectors. NK cells also produce immunomodulatory cytokines (IFN-γ) that can recruit host immune responses, perhaps contributing to a more robust anti-tumor milieu. On the other hand, NK cells cannot proliferate as massively as T cells in vivo, which might limit their ability to eradicate large tumor burdens without multiple infusions.
Target selection and resistance: Tumors may downregulate or shed the target antigen to escape CAR-T/NK. In the context of CAR-NK, even if the target is lost, the NK cell might still recognize the tumor via its innate receptors (e.g. through NKG2D ligands if the tumor is stressed from therapy). Thus CAR-NK could have a dual targeting capability: the CAR for a specific antigen and the native NK receptors for “missing-self” or stress signals. There is some evidence that residual tumor cells after CAR19-NK were still susceptible to NK killing if, for example, they lost CD19 but had low HLA or high stress ligands – whereas a CD19-negative relapse typically evades CAR-T completely. This broad recognition is an intrinsic advantage of NK cells.
In current clinical trials, CAR-NK cells have shown remarkable safety and encouraging efficacy, especially in blood cancers. However, larger studies are needed to directly compare their long-term outcomes to CAR-T. It is conceivable that for certain indications, an allogeneic CAR-NK product could be used as first-line cellular therapy due to ease of use and safety, reserving CAR-T for salvage or for cases where long-term persistence is required (like for providing immunological memory in leukemia). Combination approaches are also being considered – for example, using CAR-NK cells in conjunction with CAR-T or with post-infusion cytokine aldesleukin (IL-2) to sustain them.
In summary, genetic engineering is unlocking NK cells’ potential to be targeted “smart bombs” against cancer. CAR-NK cells merge the innate killing ability of NKs with the precision of antibody recognition. They hold promise as a platform that could overcome some limitations of CAR-T, offering an off-the-shelf, safer cell therapy. Ongoing research is optimizing CAR constructs for NK biology, exploring multiplex gene-edits to improve NK cell trafficking and resistance to suppression, and testing CAR-NKs in a variety of malignancies. The next section will delve deeper into the exciting area of stem cell-derived NK cells, which goes hand-in-hand with engineering to produce uniform NK cell products at scale.
Stem Cell–Derived NK Cells: iPSC and hESC as NK Sources
Using stem cells to produce NK cells is a transformative approach that addresses a key challenge in cell therapy – the need for a renewable, consistent cell source. Two main types of stem cells are being leveraged: hematopoietic stem cells (HSCs) (typically from cord blood or mobilized peripheral blood, as mentioned earlier) and pluripotent stem cells (either embryonic stem cells or induced pluripotent stem cells, iPSCs). The focus in recent years has been on iPSC-derived NK cells due to their versatility and scalability.
iPSC-Derived NK Cells: Induced pluripotent stem cells are generated by reprogramming adult somatic cells to a pluripotent state. They can self-renew indefinitely and differentiate into all cell lineages, including the hematopoietic lineage. Researchers have developed protocols to differentiate iPSCs into NK cells through intermediate stages that mimic embryonic hematopoiesis. Generally, an iPSC is first induced to form mesoderm, then hematopoietic progenitor cells (CD34^+CD45^+), and finally these progenitors are cultured with IL-15 and other factors to become NK cells. Feeder layers (like stromal cells expressing Notch ligands) or embryoid body co-culture systems are often used. A pioneering method by Woll et al. (2019) showed that iPSC-NK cells could be produced in feeder-free cultures with artificial nicotinamide expansion steps, yielding mature CD56^+ NK cells capable of killing leukemia.
The major advantage of iPSC-derived NK cells is the potential for an unlimited, clonal supply of starting materialj. By creating a master iPSC bank from a single donor (who could be chosen for desirable NK traits like certain KIR haplotypes), one can generate thousands of doses of NK cells that are nearly identical in phenotype and function. This overcomes donor variability and batch-to-batch differences. It also allows genetic engineering at the pluripotent stage: complex modifications can be introduced into the iPSC (using CRISPR/Cas9, for example) and the clone can be expanded, fully characterized, and selected before producing NK cells. This ensures a uniform population where every cell carries the desired edits. For instance, Fate Therapeutics has reported iPSC lines engineered with combinations of CARs, high-affinity CD16, IL-15 fusion protein, and CD38 knockout (for resistance to anti-CD38 monoclonal antibody) – after differentiation, these give rise to NK cells with multiple enhanced features for attacking myeloma. One product, FT596 (an iPSC-derived NK cell) is engineered with an anti-CD19 CAR, IL-15/IL-15Rα fusion, and a non-cleavable CD16, enabling it to kill CD19^+ malignancies and perform ADCC with any co-administered antibody. Another, FT538, includes three gene knockouts (CD38, to avoid fratricide with anti-CD38; CD52, to permit use with alemtuzumab conditioning; and iTCR to remove any residual TCR-expressing cells) and IL-15/IL-15Rα, providing an “off-the-shelf” NK for AML. These designs illustrate the power of iPSC engineering: you can pre-arm NK cells with CARs and cytokine support, and eliminate targets that a therapeutic antibody will hit or that cause self-sensitivity, all before differentiation.
From a manufacturing perspective, iPSC-derived NK cells lend themselves to large-scale production. Bioreactors can be used to differentiate millions of iPSCs into billions of NK cells in a controlled process. Because iPSCs can be expanded exponentially, there is essentially an infinite supply of starting material, limited only by bioprocessing capacity. Companies have demonstrated the ability to make cryopreserved iPSC-NK cell doses that meet release criteria for sterility, purity, identity, etc., similar to conventional cell therapy products. Another benefit is product consistency – each lot from the same clone will have the same composition (same KIR receptors, same CAR copy number, etc.), which could translate to more predictable clinical effects and easier regulatory approval as an “off-the-shelf” biological product.
Of course, there are challenges and considerations: iPSC differentiation is complex and needs to be efficient to yield high percentages of NK cells. Any undifferentiated iPSCs remaining in the product could form teratomas, a serious safety concern, so protocols include steps to eliminate or maturate away any residual pluripotent cellsf. So far, clinical trials of iPSC-derived NK cells (e.g. FT500 for advanced solid tumors, FT516 for B-cell lymphoma and leukemia, FT596 for B-cell malignancies) have reported no evidence of teratoma formation, suggesting that the differentiation and purification methods are robust. Another consideration is that iPSC lines have a fetal-like immunophenotype (HLA expression, etc.), which might cause them to be rejected by the patient’s immune system over time; however, in lymphodepleted patients this may be less of an issue, and the relatively short lifespan of NK cells might moot it. Some groups are exploring creating HLA “universal” iPSC lines (with HLA knocked out or with HLA-E overexpression to evade host NKs) to further reduce immunogenicity.
HSC-Derived NK Cells: In parallel, methods using umbilical cord blood CD34^+ cells (a more lineage-restricted stem cell source) have also been scaled up. Glycostem’s process, for example, expanded cord blood CD34 cells then induced them into NK cells over several weeks in a bioreactor with proprietary medium (GBGM) and cytokines, achieving a >1,000-fold expansion and >90% purity NK cells at the end. These NK cells (named oNKord) were given to AML patients and showed the ability to expand in vivo in some patients with minimal side effects. Celularity’s placental CD34-derived CYNK-001 has been tested in multiple indications, though detailed results are not all published. This approach is somewhat less flexible in engineering than iPSC (since CD34^+ are lineage committed and harder to gene edit extensively), but it benefits from using a more “natural” pathway of NK development. Interestingly, NK cells derived from HSCs might have longer telomeres and potentially greater proliferative potential in vivo. They also start as naive NK cells, which could allow in situ education by the patient’s environment, potentially mitigating some alloreactivity issues (though NK alloreactivity is usually beneficial in the haploidentical transplant setting).
In summary, stem cell–derived NK cells represent an exciting frontier for producing allogeneic, off-the-shelf NK cell therapies on a commercial scale. iPSC-derived NK cells in particular offer: (1) an unlimited supply from a single donor clone, (2) the ability to incorporate multiple genetic modifications at once to create “armored” NK cells, (3) batch consistency, and (4) potentially lower cost of goods when scaled, as the process can be standardized like a biologic manufacturing. The ultimate vision is an inventory of cryopreserved CAR-iNK products that can be shipped to hospitals and administered on-demand, much like a drug, but with the living cell’s ability to actively seek and destroy cancer. Ongoing trials will teach us about the efficacy and optimal dosing of these cells. If they show comparable outcomes to autologous cell therapies, this could be a paradigm shift in cell therapy – moving from bespoke patient-specific products to readily available cellular drugs.
That said, careful monitoring for any unexpected behaviors (e.g. insertional oncogenesis from integrated vectors in iPSC, or residual pluripotent cells, etc.) will be required as more patients are treated with stem cell–derived NK products. Regulatory agencies are closely evaluating these first-in-class therapies, but the early clinical experiences (no major safety flags, some promising responses) are fueling optimism that “off-the-shelf” NK cells may become a reality in the near future.
NK Cell Culturing and Manufacturing Techniques
Scaling up NK cells from laboratory research to clinical-grade doses requires specialized culture techniques and strict quality control. Unlike some cell types, NK cells can be finicky to grow and maintain. Here we discuss methods to culture NK cells at scale (including bioreactors and media choices), strategies for feeder-free and serum-free culture, and the challenges of ensuring a consistent, safe cell product.
Bioreactors and Automated Culture Systems: To produce the billions of cells often needed for cell therapy, static tissue culture flasks are insufficient. Instead, bioreactor systems and closed, automated platforms have been adopted for NK cell manufacturing. Examples include G-Rex gas-permeable static culture devices (which allow high-density culture by facilitating oxygen diffusion), wave bag bioreactors (rocking motion bioreactors that keep cells suspended and aerated), and stirred-tank bioreactors with microcarriers. Studies have shown that NK cells and other lymphocytes can achieve higher expansion rates in perfused or agitated cultures compared to static bags. The likely reasons are better nutrient and oxygen distribution and removal of waste metabolites. One academic study noted that activated NK cells had a greater than twofold expansion when cultured in a wave bioreactor versus a static culture bag over 2 weeks. Additionally, automated systems like the CliniMACS Prodigy® combine cell separation, culture, and harvest in a single closed device. The Prodigy has specific modules for NK cell enrichment (via immunomagnetic CD3 depletion and CD56 enrichment) and for expansion in a sterile bag with perfusion. Such automation reduces labor and contamination risk, and allows process standardization. NK cells can also be sensitive to shear stress, so bioreactor stirring speeds must be optimized (too vigorous can damage cells). Newer approaches involve perfusion bioreactors that continually remove spent medium and add fresh medium, keeping cells in log-phase growth. Glycostem’s large-scale process for CD34-derived NK used a bioreactor to sequentially expand progenitors then differentiate to NK, all in one unit. This indicates that multi-step culture can be handled in automated fashion.
Feeder-Free vs Feeder-Based Cultures: As discussed, many NK expansions rely on feeder cells like K562 variants. However, using feeder cells in a GMP process raises additional regulatory hurdles – feeder cells are often of tumor origin (risk of transfer of material, although irradiation mitigates proliferation, and cells are usually washed out before final product). There is interest in feeder-free culture methods to simplify the product. Approaches include using artificial beads coated with 4-1BBL and other ligands in combination with high-dose cytokines to mimic the feeder stimulation. Another is using gene-modified stimulatory cell lines that are themselves inert (for example, clone cells that can be easily depleted at the end). Nonetheless, a number of current clinical processes still use irradiated K562 feeders because they yield the best expansion. A compromise technique uses feeder cells that are lethally irradiated and then encapsulated or fixed, so they provide stimulation but cannot mix with the final harvest easily. Feeder-free protocols often require more intensive cytokine use and can result in different phenotypes (some studies show feeder-expanded NKs have higher activation receptor expression and greater cytotoxicity than solely cytokine-expanded NKs).
Culture Media and Supplements: NK cells have specific media requirements. Commonly used base media include RPMI-1640 (with supplements) and alpha-MEM for NK-92, as well as specialized formulations. For clinical manufacturing, several GMP-grade media are popular: CellGro/SCGM (a serum-free medium optimized for lymphocyte growth), X-VIVO 10 or 20 (serum-free media from Lonza, often used for T/NK cell culture), and NK MACS® medium by Miltenyij. Each organization often develops its own proprietary medium tweaks. A critical factor is whether to use human serum or other protein supplements. NK cells typically grow better with some form of serum or albumin present. Fetal bovine serum (FBS) has traditionally been used at 5–10%, but it carries risks: potential prion or virus transmission, and it introduces animal proteins that could be immunogenicj. Regulatory guidance strongly encourages eliminating animal-derived components for cell therapy manufacturing. Many clinical protocols have switched to human AB serum (human platelet lysate is another alternative), which provides necessary growth factors with less risk. Human AB serum (pooled from screened donors) at 5–10% can support NK expansion well. Still, sourcing large quantities of AB serum is a challenge as cell therapy trials scale up. There is also batch variability in serum which can affect cell yields and phenotype. Therefore, companies like Glycostem have invested in serum-free media development. Glycostem’s GBGM medium enabled a 4-log expansion of NK cells from CD34+ without any serum. Other groups use cocktails of recombinant albumin, insulin, transferrin, etc., to replace serum. Some products (e.g. Acepodia’s oNK cells) use platelet lysate as a xeno-free supplement, which is rich in growth factors and can substitute for FBS. Achieving completely serum-free, feeder-free NK expansion is somewhat the “holy grail” for NK manufacturing to maximize reproducibility and safety. Progress has been made – for instance, one trial expanded peripheral blood NK ~3000-fold in 18 days using a defined serum-free medium with IL-2, anti-CD3 (to stimulate accessory cells), and heparin, achieving clinical-scale yields. The presence of heparin in that protocol likely helped by preventing cell aggregation and possibly modulating IL-2 availability.
Scalability and Yield Challenges: NK cells have a tendency to undergo activation-induced cell death if overstimulated, and not all NK donors expand equally (there’s donor variability in expansion potential). This can make it hard to predict yields. To mitigate this, some processes include a “rest” phase or carefully controlled cytokine levels rather than continuous high stimulation. Additionally, NK cells may require a certain level of feeder ratio to initiate expansion; in large volumes, maintaining sufficient contact between NKs and feeders or beads can be tricky. Stirred systems that keep cells well-mixed help. Another challenge is cryopreservation: NK cells can be frozen and thawed, but some loss of function (particularly in primary NKs) is noted after thaw. Formulation in 5–10% DMSO and albumin-containing cryomedium is standard. Interestingly, some studies show that CAR-NK cells (especially those with IL-15 support) survive thawing better. Ensuring that the final product retains high viability post-thaw (typically >70% viability is required) and retains cytotoxic function is part of QC.
Quality Control (QC) Considerations: A clinical NK cell product must meet stringent criteria before release for infusion:
Identity/Purity: The product should contain the intended cells – typically defined as % CD56^+CD3^- cells. Many protocols aim for >85% purity NK cells. Residual T cells must be very low (to avoid GvHD in allogeneic products). Often a specification like “CD3^+ < 1%” or “<1×10^5 residual T cells per dose” is set. B cell contamination should also be minimal to avoid transferring host EBV or tumor cells. In CAR-NK, identity also includes % of cells that express the CAR (transduction efficiency).
Viability: Usually >80% viable by Trypan blue or 7-AAD staining is required.
Sterility and Mycoplasma: The product must be free of bacterial/fungal contamination and mycoplasma. Given the short shelf-life of cell products, release often uses rapid sterility assays or in-process monitoring to ensure no contamination.
Endotoxin: Should be below a certain threshold (often <5 EU/mL) since media and reagents could introduce endotoxins.
Potency: This is important but tricky – a cell therapy needs a defined potency assay demonstrating its biological activity. For NK cells, a common potency assay is an in vitro cytotoxicity assay against a target cell line (for example, % lysis of K562 leukemia cells at a certain effector:target ratio). Some use a CD107a degranulation assay or IFN-γ production as a surrogate. Regulators expect that each batch of cells shows a minimum level of cytotoxic function. For CAR-NK, the potency assay might be lysis of antigen-positive target (e.g., CD19^+ tumor cells for a CD19 CAR-NK).
Genetic stability: If cells have been expanded long-term (especially relevant for iPSC or cell lines), karyotype or copy number variation analysis is sometimes done to ensure no malignant transformation or gross chromosomal abnormalities. NK-92 lines, for example, should be periodically checked that they remain identical to the starting bank.
Phenotypic characterization: Apart from purity markers, additional characterization like checking KIR expression, activation markers (NKG2D, DNAM-1 levels), or exhaustion markers can be done for information, though not necessarily as release criteria. For engineered cells, confirming the expression of the transgene (CAR expression level) via flow cytometry is key.
Manufacturing processes also incorporate safeguards like sampling for replication-competent virus if viral vectors were used, since any replication-competent retrovirus (RCR) must be undetectable.
Scale and Cost: The field is pushing towards manufacturing processes that can produce on the order of 10^10 NK cells in a single batch, which could yield 50–100 doses. This is already being attempted in large bioreactors for allogeneic products. The cost of goods for NK cell therapy is currently high (due to cytokines like IL-15, media, labor, etc.), but there’s optimism it will be less than autologous CAR-T if centralized manufacturing and economies of scale are realized (no need for separate product per patient). The removal of feeder cells and serum, if achieved, will also streamline production and reduce QC testing burden (since each additional component like human serum needs its own testing and qualification).
In conclusion, the cultivation of NK cells for therapy is a sophisticated process requiring careful control of the environment to maximize yield and function. Bioreactors and automation are enabling larger-scale production with reproducibility, while serum-free and feeder-free approaches are improving the safety and consistency of the final product. Quality control measures ensure that what is given to patients is a pure population of highly cytotoxic NK cells with minimal impurities or risks. As NK cell therapies advance through clinical trials, continued innovation in manufacturing is expected to improve efficiency – for example, the integration of upstream NK expansion with downstream formulation in closed systems, or the use of novel expansion stimuli (small molecules that promote NK metabolic fitness, etc.). These technologies will be critical to bring NK cell therapies to mainstream clinical use, where hundreds or thousands of doses might be needed for larger patient populations.
Combination Therapies Involving NK Cells
Combining NK cell therapy with other treatments can yield synergistic effects, leveraging multiple modes of attack on the tumor. NK cells naturally interact with antibodies and cytokines, and they are subject to immune checkpoints just like T cells. Therefore, rational combinations – with monoclonal antibodies, immunomodulators, or conventional chemo/radiotherapy – are being actively explored to enhance NK cell efficacy in the clinic.
Monoclonal Antibodies (ADCC Enhancement): One of the most straightforward combinations is NK cell therapy with tumor-specific antibodies that engage NK cells via ADCC. Many FDA-approved therapeutic antibodies (rituximab for CD20^+ lymphoma, trastuzumab for HER2^+ breast cancer, cetuximab for EGFR^+ colorectal/head&neck cancer, etc.) rely in part on NK cell-mediated ADCC for their efficacy. Infusing additional NK cells (especially allogeneic NKs with high CD16 levels) into patients receiving these antibodies can intensify the ADCC effect. For example, a Phase II trial in advanced head and neck cancer combined cetuximab (anti-EGFR mAb) with infusions of ex vivo expanded haploidentical NK cells. The group receiving NK cells + cetuximab showed improved progression-free and overall survival compared to cetuximab alone. Specifically, median PFS was extended to 6 months versus 4.5 months with antibody alone, and similarly OS improved (though patient numbers were small). Another clinical example is the AFM13 trial: AFM13 is a bispecific engager that binds CD30 on lymphoma cells and CD16 on NK cells. Cord blood-derived NK cells were pre-complexed with AFM13 and given to Hodgkin lymphoma patients, resulting in an 89% objective response rate in a Phase I study – a striking result in a refractory population, attributable to potent ADCC by the adoptively transferred NK cells engaging through the bispecific antibody. Even without engineered engagers, endogenous CD16 on NK cells makes them natural partners for any IgG1 therapeutic antibody. High-affinity CD16 variants: About 15% of humans carry the high-affinity 158V allele of CD16 which binds IgG with higher affinity and mediates stronger ADCC. Some adoptive NK strategies select donors with the 158V/V genotype or even engineer NK cells to express a high-affinity CD16 (for instance, one group electroporated mRNA for CD16 158V into NK cells to enhance rituximab activity). Additionally, there are attempts to prevent the shedding of CD16 from NK cells upon activation (since ADAM17 can cleave CD16); for example, a non-cleavable CD16a was engineered into an iPSC-NK product to ensure sustained ADCC capability. Overall, pairing NK cells with monoclonal antibodies (sometimes called ADCC combos) is a logical way to exploit NK biology. It is especially attractive for diseases like lymphoma, where an antibody (rituximab) is standard of care – adding NK infusions could convert partial responders to complete responders. One challenge is that patient serum contains IgG which can occupy CD16 (IVIG, or even the therapeutic antibody itself can saturate NK cell Fc receptors if dosing is high); scheduling NK infusions at a time when antibody levels produce optimal opsonization but not saturation is something to consider.
Immune Checkpoint Blockade: Checkpoint inhibitor drugs (anti-PD-1, anti-PD-L1, anti-CTLA-4, etc.) have revolutionized T cell-based immunity against cancer. NK cells too are subject to checkpoints. They express PD-1 in certain tumor microenvironments (especially “exhausted” NK cells in chronic stimulation scenarios can upregulate PD-1) and their activity can be suppressed by tumor PD-L1. Therefore, PD-1 or PD-L1 blocking antibodies can indirectly or directly enhance NK cell function. For instance, pembrolizumab (anti-PD-1) may reinvigorate intra-tumoral NK cells that express PD-1, and anti-PD-L1 (e.g. atezolizumab) can prevent tumor PD-L1 from engaging PD-1 on both T and NK cells. Interestingly, PD-L1 antibodies can also arm NK cells through ADCC: when NK cells encounter a tumor cell coated with an anti-PD-L1 antibody, they can kill that cell via CD16 recognition of the antibody’s Fc. Durvalumab and avelumab (both anti-PD-L1) have been noted to have this dual function – blocking PD-1/PD-L1 interaction and simultaneously marking the tumor for NK killing. Another checkpoint relevant to NK is NKG2A. NKG2A is an inhibitory receptor on NK cells (and some T cells) that binds HLA-E. Tumors often upregulate HLA-E as a way to inhibit NK cells (as HLA-E signals “self” through NKG2A). The drug monalizumab is a monoclonal antibody that blocks NKG2A, thereby freeing NK cells (and NKG2A^+ CD8 T cells) from that inhibition. In a Phase II trial for head and neck cancer, combining monalizumab with cetuximab resulted in a higher response rate than cetuximab alone, presumably due to enhanced NK (and T) activity when NKG2A is blocked – HLA-E on tumors could no longer protect them from NKG2A^+ NK cells. Similarly, anti-KIR antibodies (like lirilumab, which blocks KIR2DL1/2/3) were tested to prevent inhibitory KIRs from seeing their HLA-C ligands. Lirilumab alone did not show strong clinical efficacy, likely because blocking one set of KIRs is not sufficient or tumors had other escape, but it is being tried in combination with other immunotherapies. Additionally, NK cells express checkpoints like TIGIT, TIM-3, LAG-3 in tumor settings. TIGIT especially is an inhibitory receptor on NK (and T cells) that binds PVR (CD155) on tumor cells. Anti-TIGIT therapies in development (e.g. tiragolumab) might enhance NK cell-mediated immunity – indeed, preclinical data showed TIGIT blockade can invigorate NK cells and improve anti-tumor responses. In summary, checkpoint blockade can be a powerful adjunct to NK cell therapy, by unleashing NK cells’ activity in the tumor microenvironment. A practical example: if a patient receives an NK cell infusion, giving an anti-PD-1 antibody could help those NK cells (and endogenous T cells) function better in tumors with high PD-L1. Checkpoints specific to NK, like NKG2A and KIR, are promising targets to pair with NK therapies. Ongoing trials are combining anti-NKG2A or anti-PD-1 with NK cell infusions in refractory acute myeloid leukemia and solid tumors.
Cytokine and Immune Modulator Combinations: NK cells respond to certain cytokines in vivo. High-dose IL-2 was historically given after NK infusion in some trials (e.g. Miller et al. in 2005 gave IL-2 to patients after haploidentical NK infusion). IL-2 helps the transferred NK cells proliferate and survive, but IL-2 can also expand Tregs which is counterproductive. Newer approaches use IL-15 instead. IL-15 can be given as an infusion (complexed with IL-15Rα to increase half-life, e.g. ALT-803, also called N-803, a superagonist IL-15). ALT-803 has been combined with NK cell therapy to sustain the NK cells in vivo and showed some success in early trials (e.g. in solid tumors). Another modulator is lenalidomide, an immunomodulatory drug that can activate NK cells indirectly by reducing inhibition from cereblon pathway in tumor cells and by downmodulating NK cell ligands. Low-dose lenalidomide has been combined with NK cell therapy in a trial for multiple myeloma, aiming to exploit lenalidomide’s known ability to boost NK cell cytotoxicity (lenalidomide can increase NK cell synapse formation and IL-2 production by T cells). Similarly, haploidentical stem cell transplantation is sometimes augmented by post-transplant NK cell infusions to speed immune reconstitution and graft-versus-leukemia effect. In that scenario, patients often receive IL-15 agonists or low-dose IL-2 to support the NK cells post-transplant.
Chemo and Radiation Sensitization: Certain chemotherapies and radiation therapies can make tumor cells more vulnerable to NK cell killing. For example, many DNA-damaging chemotherapeutics (like 5-FU, doxorubicin, melphalan) have been shown to upregulate NKG2D ligands on tumor cells. This is because the cellular stress or DNA damage response triggers pathways that lead to expression of MICA/B and ULBPs on the cell surface. When these ligands are up, NK cells more readily recognize and kill the tumor. A specific demonstration: irradiating tumor cells can induce heat-shock proteins and upregulate MICA/B; an experimental study found that sublethal heat shock or ionizing radiation increased NKG2D ligand expression on various carcinoma cell lines and consequently their susceptibility to NK cell cytotoxicity. The peak of ligand induction was a few hours after treatment, aligning with when NK cell killing was most effective. Thus, combining localized radiation (to prime tumors) followed shortly by NK cell therapy might produce better tumor control than either alone. Some clinical protocols in development use low-dose radiation on a tumor site to draw NK cells in and make the cancer cells more “visible” to them. Chemotherapy preconditioning is already a part of most adoptive cell therapy regimens: patients receive lymphodepleting chemo (like cyclophosphamide + fludarabine) before cell infusion. Besides creating “space” and homeostatic cytokines for the infused cells, this regimen can reduce immunosuppressive cells and possibly upregulate stress ligands on residual tumor cells. The 2005 AML NK trial attributed part of its success to the high-dose cyclophosphamide/fludarabine given, which not only cleared patient lymphocytes (preventing rejection of donor NKs) but also might have made the leukemia cells more susceptible by reducing their expression of HLA and increasing activating ligand expression. There is also evidence that some chemotherapies like bortezomib (proteasome inhibitor) upregulate TRAIL death receptors on tumor cells, which NK cells can then exploit via TRAIL-mediated killing. Another angle is that chemotherapy can alter the tumor microenvironment: for instance, platinum chemotherapy can induce immunogenic cell death releasing ATP and HMGB1, leading to dendritic cell activation that could secondarily enhance NK cell activity; or deplete myeloid-derived suppressor cells that were inhibiting NK cells.
NK Cells with Other Cell Therapies: Combining NK cells with T cell-based therapies is an area of interest too. One idea is using NK cells to bridge the time until CAR-T cells (which take weeks to manufacture) are ready; an off-the-shelf NK could be given immediately to debulk disease. Another concept is sequential or concomitant CAR-T and CAR-NK, possibly targeting different antigens to mitigate escape. There's even a trial combining allogeneic NK cells with tumor-infiltrating lymphocytes (TILs) in melanoma, reasoning that NK cells can kill MHC-loss variants while TILs handle MHC-positive tumor cells.
Results in Clinical Trials: Many of these combination strategies are in early-phase trials. To highlight a few: in acute myeloid leukemia, a pilot study added NK cell infusions to haplo transplant plus IL-15 (ALT-803) and showed improved leukemia-free survival compared to historical controls. In multiple myeloma, NK cells combined with elotuzumab (anti-SLAMF7 mAb) and IMiDs yielded some deep responses. In solid tumors like metastatic colorectal cancer, a study combined NK cells with cetuximab and NKG2A blockade (monalizumab) – capitalizing on all three: ADCC, checkpoint release, and NK infusion – with early signs of activity. It should be noted that some combinations might increase toxicity; for instance, IL-2 with NK cells can cause capillary leak syndrome, and adding NK cells to a PD-1 inhibitor could (in theory) add to autoimmune side effects if any. So far, however, NK therapy combos have been well-tolerated.
In summary, combination therapy is likely to be key for maximizing NK cells’ therapeutic potential. Monotherapies with NK cells have shown modest efficacy in certain advanced cancers, but by pairing NK cells with complementary agents, one can address multiple mechanisms: target cell opsonization (mAbs), tumor microenvironment suppression (checkpoint inhibitors), and even direct NK activation (cytokines). As an example of a multifaceted approach, one could envision a regimen for refractory lung cancer: low-dose radiation to tumor sites (increase NK ligands), followed by allogeneic CAR-NK cells targeting, say, PD-L1 on tumor (for direct kill and ADCC), combined with an anti-NKG2A to ensure NK cells aren’t blocked by HLA-E, plus an IL-15 superagonist to sustain the NK cells. While complex, such a regimen targets the tumor from many angles at once. Ongoing and future trials will inform which combinations yield the most synergistic benefit with acceptable safety. The flexibility and safety profile of NK cells makes them attractive to pair with other therapies – they can be given multiple times and from allogeneic sources, so one can integrate them into standard treatment schedules more easily than, say, autologous CAR-T. The coming years will likely see NK cells incorporated as an adjunct to existing cancer therapies, potentially improving outcomes in diseases like acute leukemia (with post-remission NK infusions to prevent relapse), solid tumors (converting partial responses to durable remissions), and even viral infections (e.g. using cytokine-activated NK cells alongside antivirals in refractory viral diseases).
Clinical Trial Landscape and Efficacy of NK Cell Therapies
NK cell therapies have progressed from early experimental trials to numerous ongoing clinical studies across cancer types. The landscape as of 2025 includes a mixture of academic-led trials and industry-sponsored trials evaluating both unmodified NK cells and engineered NK products (CAR-NK, bispecific-engager-armed NKs, etc.). Here we provide an overview of key clinical trial findings, organized by disease category, and discuss safety and efficacy outcomes that have been reported.
Hematologic Malignancies – Leukemias and Lymphomas: Some of the first successful NK therapy reports came in acute myeloid leukemia (AML). In 2002, Ruggeri et al. showed that in haploidentical stem cell transplantation for AML, if the donor’s NK cells were alloreactive (KIR mismatched), patients had significantly lower leukemia relapse rates. This finding, though in the transplant context, highlighted NK cells’ graft-versus-leukemia effect. In 2005, Miller and colleagues conducted a Phase I trial infusing haploidentical NK cells into patients with refractory AML after lymphodepleting chemotherapy (cyclophosphamide and fludarabine). The results were remarkable for that time: 5 of 9 poor-prognosis AML patients achieved complete remission after NK infusion. Notably, there was no GvHD observed, and toxicity was limited to mild infusion reactions and IL-2 side effects (patients received IL-2 to support NK survival). This provided a proof-of-concept that NK cells can induce remissions in acute leukemia with minimal toxicity. Subsequent AML trials (mostly Phase I/II) have explored different NK sources – e.g. cord blood NK cells, ex vivo expanded donor NKs, or “memory-like” cytokine-preactivated NKs. A recent trial from Washington University used IL-12/15/18 preactivated “memory-like” NK cells in AML and reported durable remissions in several patients with relapsed AML, with some responses lasting over a year. These NK cells expanded in vivo and exhibited an activated phenotype. For maintenance therapy in AML, MD Anderson tested repeated infusions of allogeneic NK cells after chemotherapy and saw hints of improved relapse-free survival in high-risk AML, although controlled Phase III data are not yet available.
In B-cell lymphomas and leukemias, CD19-targeted CAR-NK cells have gained a lot of attention. The first-in-human trial by Liu et al. (published 2020) treated 11 patients with CD19-positive cancers (non-Hodgkin lymphoma or CLL) with cord blood-derived CAR-NK cells expressing anti-CD19 CAR, IL-15, and iC9 safety switch. The CAR-NK cells were given without cytokine support and without causing CRS. The outcomes: 8 of 11 patients responded, including 7 complete remissions, and these responses occurred rapidly (within 30 days). Although the follow-up was short, it was notable that the NK cells did not permanently engraft – they were mostly undetectable by day 30–60, yet some remissions were maintained for months, suggesting perhaps the patients’ immune system could keep the cancer in check after the NK “push”. No patient developed grade 2+ CRS, neurotoxicity, or GvHD, highlighting the favorable safety. Building on that, other trials have emerged: for example, a CD19 CAR-NK product (NKX019 by Nkarta) was tested in relapsed B-cell lymphoma. In interim results, 7 of 10 patients at the higher dose achieved complete responses (CR). This ~70% CR rate is on par with autologous CAR-T therapies in similar populations, which is very encouraging. Another study (Houston Methodist/Lei et al. 2023) used a cord blood CAR-NK with 4-1BBζ and IL-15, giving three fractionated doses to B-cell lymphoma patients. They reported an overall response rate of 63% and CR rate of 50% at 1 month, even at relatively low cell doses, and again no CRS or ICANS observed. Some patients had durable remissions ~1 year with no maintenance, others relapsed within months, possibly correlating with the extent of CAR-NK persistence. Combination of CAR-NK with antibodies is also in trials: one trial pairs CD19 CAR-NK with rituximab in aggressive lymphoma (to see if the NK can also mediate rituximab ADCC against any CD19-negative, CD20-positive tumor cells).
Multiple myeloma (MM) has seen NK trial activity too. BCMA-CAR NK cells are in early trials for MM, including an iPSC-derived FT576 mentioned earlier which has anti-BCMA CAR, IL-15 and CD16. Data are pending, but preclinical results showed these cells could overcome MM resistance mechanisms. Another approach in MM was using donor NK cells after autologous stem cell transplant as consolidation – a Phase II trial did this with IL-2 DIPHERG (to support NK) but results did not show a clear benefit. However, when using adaptive NK cells (from certain donor selection with KIR-ligand mismatch), there is evidence of graft-vs-myeloma effect. A notable trial in progress is using NK cells with daratumumab (anti-CD38 mAb) after transplant, but since daratumumab can bind CD38 on NK cells and fratricide them, clever engineering (like CD38-knockout NK cells) is being applied in product design.
Solid Tumors: Historically, NK cell therapy in solid tumors has faced more hurdles – solid tumors are adept at creating a suppressive microenvironment, and NK cell homing to solid tumors is less efficient. Early trials of autologous NK cells in melanoma, RCC, colorectal cancer in the 1990s and 2000s showed safety but little efficacy. For instance, in renal cell carcinoma (RCC) and metastatic melanoma, autologous NK infusions did not yield significant tumor regression, likely due to tumor expression of self MHC and other escape mechanisms. However, newer trials with allogeneic, expanded NKs and combination treatments have reported some success. A pilot trial in refractory non-small cell lung cancer combined haplo NK cell infusions with chemotherapy; a few patients had partial responses, and interestingly the NK cells were found in tumor biopsies post-infusion, indicating tumor infiltration. Another case series in colorectal carcinoma observed that intra-arterial infusion of allogeneic NK cells into liver metastases led to necrosis in some lesions, pointing to possible efficacy when NK cells can be delivered directly to tumor sites.
One of the most promising solid tumor NK trials to date was in Merkel cell carcinoma (an immunogenic skin cancer). A trial of an anti-PD-1 plus allogeneic NK cells reported a CR in a patient who had failed checkpoint therapy before – suggesting the NK cells provided the missing effect (this is anecdotal but encouraging). Similarly, in pediatric neuroblastoma, allogeneic NK cells have been combined with the anti-GD2 antibody dinutuximab as a post-transplant consolidation. A Phase II study (NCT01576692) using haplo NK + dinutuximab in high-risk neuroblastoma showed improved 2-year event-free survival compared to historical controls (though not randomized). This combination makes sense as GD2 antibody triggers NK ADCC, and adding more NKs likely amplifies tumor killing.
Cytokine-Induced Memory-Like (CIML) NK cells in solid tumors: While strong results were seen in AML, in solid tumors they may need help from checkpoints or cytokines. Trials are ongoing in head & neck cancer combining CIML NK cells with cetuximab and NKG2A blockade, which is an all-out attempt to drive NK activity in the tumor.
Safety in Trials: Across the hundreds of patients treated on NK trials so far, the safety profile has been very favorable. CRS (cytokine release syndrome) is rare; when it occurs, it’s usually grade 1–2 (fever, mild hypotension) and often attributed to concomitant IL-2 rather than NK cells themselves. For example, in Miller’s AML trial, some patients had low-grade fevers/chills after NK infusion, likely due to IL-2 injections. In the MD Anderson CAR-NK trial, they explicitly reported no CRS or neurotoxicity at any dose. Neurotoxicity (ICANS), commonly seen with CAR-T, has essentially not been observed with CAR-NK or unmodified NK infusions. Graft-versus-host disease has not occurred even with using related donor or third-party NK cells, consistent with the biology that NK cells don’t cause GvHD. Some patients, especially those getting IL-2, have experienced transient lymphopenia, liver enzyme elevation, or lung infiltrates (could be related to cytokine side effects). But compared to T cell therapies, the adverse event spectrum is mild.
One notable manageable toxicity is “Infusion reactions” – chills, fever during the NK infusion, which is not uncommon but typically grade 1. Premedication with acetaminophen and antihistamines is often done. If DMSO-cryopreserved products are used, DMSO can cause some patients to cough or get a bad taste.
In a few studies where high doses of NK were given after transplant, some cytopenias were seen (hard to disentangle from chemo effects). There was also a theoretical concern that allogeneic NK cells could reject the host (so-called “graft-versus-host in the absence of T cells” – better called engraftment syndrome), but no serious cases reported. Allogeneic NK cells do not persist long enough to cause lasting issues; in fact, a challenge is they often require multiple infusions to maintain a presence.
Efficacy Summary: The efficacy signals vary by disease context:
AML: roughly 30–50% complete remission rates in relapsed/refractory AML across various NK approaches, which is significant in such a tough setting. Memory-like NK cells reported ~50% CR in relapsed AML in one trial. These responses, though, sometimes are short-lived unless consolidated (some patients eventually relapsed).
B-cell malignancies: CAR-NK cells targeting CD19 have achieved CRs in ~60–70% of patients in early trials, which approaches the efficacy of CAR-T (where CR rates are ~70–80% in similar groups). The durability is still under follow-up; in one study median PFS was ~9.5 months which is encouraging for a Phase I. Non-engineered NK with antibodies also show improvement in response rates (e.g. NK + rituximab vs rituximab alone).
Multiple Myeloma: not many published results yet; however, one study of NK cells with elotuzumab (anti-SLAMF7) in lenalidomide-refractory myeloma showed a couple partial responses. More data will come from BCMA CAR-NK trials.
Solid tumors: here the efficacy has been modest on its own, but combinations are yielding better outcomes. It’s fair to say no solid tumor has yet had a breakthrough NK therapy success like the CAR-T in B-ALL. For example, in metastatic melanoma, a phase II of autologous NK + IL-2 failed to meet endpoints. But newer work: the AFM13 + NK in Hodgkin’s (which is technically a solid tumor in that it’s in nodes) had 1 CR and 11 PRs out of 13 patients – extremely high response rate (though needs durability data). In liver cancer (HCC), a trial of allogeneic NK after TACE (trans-arterial chemoembolization) suggested improved tumor necrosis vs TACE alone, but survival data are pending.
The clinical trial landscape is rapidly expanding. As of early 2024, over 100 trials involving CAR-NK cells alone were registered, and hundreds more with non-engineered NK cells. Companies like Takeda, MD Anderson (in partnership with Takeda), Nkarta, Fate Therapeutics, MD Biomedical, and others have multiple Phase I/II studies ongoing. Indications range from AML, ALL, NHL, CLL to solid tumors like GBM (a trial of EGFR-CAR NK for glioblastoma is ongoing), lung cancer, gastrointestinal cancers, and even virus-associated malignancies (CAR-NK targeting EBV antigens for NK/T-cell lymphoma, for example). There is also at least one trial using NK cells in severe COVID-19 (aiming to clear infected cells by missing-self targeting).
Endpoints being measured include overall response rate (ORR), complete remission (CR) rate, duration of response (DoR), progression-free survival (PFS), and overall survival (OS). Many early trials focus on ORR and CR as primary endpoints to gauge efficacy. For example, in lymphoma trials, day 30 ORR is often reported. In transplant settings, endpoints might be relapse rate at 6 months, or graft vs host-free, relapse-free survival (GRFS). So far, the improvement in relapse prevention post-transplant with NK cells is still being studied (a Phase II trial of haplo NK infusion after haplo transplant in AML is ongoing to see if it lowers relapse vs historical rates).
The consistency of safety findings (lack of severe toxicity) stands out across the landscape. This may expedite regulatory pathways, as safety issues often slow development of cell therapies. The first approvals of NK cell therapies may happen in the next couple of years if Phase II trials confirm efficacy signals.
For instance, if the CD19 CAR-NK results hold in a larger cohort, one could envision an approval for relapsed CD19^+ lymphoma as an off-the-shelf cell therapy, especially for patients who cannot wait for CAR-T or who failed CAR-T. Another candidate is cord blood-derived NK cells combined with monoclonal antibody for acute leukemia minimal residual disease – if shown to reduce relapse, it could become standard as consolidation.
In summary, clinical trials have established that NK cell therapy is feasible and safe, with proof-of-concept efficacy in multiple settings including AML, lymphomas, and as adjuncts to antibodies. We are seeing “generation 1” NK therapies delivering encouraging results – complete remissions in heavily pretreated patients, which is remarkable given these are often donor-derived cells that do not permanently engraft. The challenge remains to improve the durability of responses. Approaches like gene-editing NK cells for persistence (e.g. mbIL-15 expression) and giving multiple doses are being trialed to address this. If durability can be improved, NK cell therapies could potentially cure a subset of patients or at least bridge them to transplant or other definitive therapies. Even in their current form, NK therapies can fill niches: for example, as a safer alternative to CAR-T in older patients or those with co-morbidities where CAR-T toxicity is a concern.
The clinical data also provide valuable lessons: the importance of preconditioning (most successes used lymphodepletion prior to NK infusion to allow NK expansion), the utility of combination therapy (e.g. NK + antibody yields better results than either alone), and confirmation that allogeneic NK cells can be given without HLA matching (no GvHD) which greatly simplifies logistics. As more Phase II/III trials read out, we will gain clarity on where NK cell therapy will fit in treatment algorithms. It’s conceivable that NK cells might be used earlier in disease (e.g. first remission AML to eradicate minimal residual disease, or frontline with antibodies in lymphomas), rather than as a last resort.
Finally, a notable aspect of NK cell trials is that many are academic-industrial collaborations – the field is benefitting from both the agility of academia (to try new donor sources, new combinations) and the resources of industry (to produce engineered NK products under GMP and run multicenter trials). This synergy will likely accelerate progress.
In conclusion, the clinical trial landscape for NK cell therapy is dynamic and rapidly evolving. With each trial, our understanding of optimal practices (dosing, scheduling, gene modifications, combination agents) deepens. The results to date already demonstrate that NK cells can mediate meaningful clinical responses in some of the most challenging cancers, doing so with minimal toxicity. This sets the stage for NK cell-based treatments to become an important component of immuno-oncology, either as stand-alone cell therapies or integrated into combination regimens. The coming years will determine how broadly effective NK therapies can be and whether they can achieve the high cure rates in cancer that we aspire to. But the progress so far, as detailed in this review, firmly establishes natural killer cells as a powerful and versatile weapon in the therapeutic arsenal – one that is moving swiftly from bench to bedside with much promise for patients.
Natural Killer cells, once considered only the innate immune system’s rapid responders, have now emerged as a highly adaptable platform for cellular therapy. In this review, we have detailed how NK cells develop and function, emphasizing their unique biology – from the balance of activating/inhibitory signals that govern their cytotoxicity, to their armamentarium of perforin/granzyme, death ligands, and cytokines. This intrinsic ability to distinguish stressed or “missing-self” cells and kill them swiftly makes NK cells a natural candidate for cancer immunotherapy and antiviral applications.
Adoptive transfer of NK cells has progressed significantly over the past two decades. Protocols for isolating and expanding NK cells from various sources (peripheral blood, cord blood, cell lines, iPSCs) now allow the generation of clinical-grade NK cell doses in the billions, with automated bioreactors and serum-free media improving scalability. Early clinical studies established that NK cell infusions – even from partially HLA-mismatched donors – are safe, not causing GvHD, and can produce remissions in leukemia patients. The lack of severe cytokine release or neurotoxicity in trials to date is a recurring theme, even as NK cell therapies become more potent via genetic engineering. This favorable safety profile may allow NK cell treatments to be administered in settings (and patient populations) where CAR-T cells or intense T cell therapies would be too toxic.
Conclusion
The advent of CAR-NK cells and other engineered NK products has opened new possibilities. We are now able to custom-design NK cells with targeted specificity (CARs for tumor antigens), improved persistence (IL-15 secreting “armored” NKs), and resistance to inhibition (via checkpoint receptor knockouts), as well as use pluripotent stem cells to mass-produce uniform NK cell therapies. These sophisticated cellular products are being tested in clinical trials with very promising initial results, particularly in hematologic malignancies where response rates with CAR-NK cells mirror those of CAR-T cells. It is noteworthy that in head-to-head context, CAR-NK cells have shown similar efficacy in clearing B-cell tumors but with dramatically reduced side effects. If these results are confirmed in larger trials, CAR-NK therapies could become an off-the-shelf alternative or complement to patient-specific CAR-T therapy, potentially transforming the cell therapy manufacturing paradigm and improving access.
We have also highlighted the versatility of NK cells in combination strategies. By working in concert with monoclonal antibodies, NK cells amplify ADCC, as evidenced by improved outcomes when NK infusions are added to antibody therapy in clinical studies. Checkpoint blockade and cytokine support can further unleash NK cells in the tumor microenvironment, addressing two major challenges in solid tumor immunotherapy: the immunosuppressive milieu and poor immune cell infiltration. Meanwhile, the ability of certain conventional treatments (chemo/radiation) to enhance NK-recognizable stress ligands provides a rationale for integrating NK cells into multimodal therapy regimens.
The clinical trial landscape for NK cell therapy, as of 2025, is rich and rapidly expanding. Hundreds of trials are exploring NK cells against a spectrum of cancers – from acute leukemias and lymphomas, to solid tumors such as glioblastoma, lung, breast, and gastrointestinal cancers. In many of these early-phase studies, NK therapies are achieving robust response rates with minimal toxicity, validating decades of preclinical work and generating excitement for larger trials. Key questions moving forward include: How durable are these NK cell-induced remissions? Will strategies like repeated dosing or in vivo cytokine support be needed to maintain long-term control? Can engineering strategies (like those used in iPSC-NK cells) overcome the persistence and homing limitations inherently seen with NK cells? Ongoing phase II/III trials and correlative science will shed light on these issues. Moreover, manufacturing and logistical challenges – while still non-trivial – are being surmounted, with the prospect that NK cell products could be stored and delivered on-demand to treatment centers, streamlining what has historically been a cumbersome process for autologous cell therapies.
In conclusion, Natural Killer cells have transitioned from basic immunologic curiosity to a versatile therapeutic modality at the forefront of immunotherapy research. They bring together desirable features of innate and adaptive immunity: an ability to rapidly kill diverse targets without prior sensitization, and now through engineering, an ability to specifically seek out tumor antigens and persist longer. With continued innovation, NK cell-based treatments may achieve outcomes rivaling or surpassing existing cell therapies, expanding the arsenal of weapons we have to fight cancer and other diseases. The coming era of “off-the-shelf” cell therapies will likely feature NK cells prominently – living drugs that can be delivered to patients with the ease of a pharmaceutical, yet with the intelligence of a cytotoxic lymphocyte that can hunt down and eliminate malignant cells. The success seen so far in clinical trials sets a strong foundation for the broader application of NK cell therapies. As our understanding and technology advance, NK cells are poised to play a central role in next-generation immunotherapies, fulfilling their eponymous potential as the body’s own natural killers and as powerful allies in the clinic against life-threatening diseases.
References
Caligiuri MA. Human natural killer cells. Blood. 2008;112(3):461–469.
Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9(5):503–510.
Romee R, Leong JW, Fehniger TA. Utilizing cytokines to function-enable human NK cells for the immunotherapy of cancer. Scientifica (Cairo). 2014;2014:205796.
Liu E, Marin D, Banerjee P, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382(6):545–553.
André P, Denis C, Soulas C, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell. 2018;175(7):1731–1743.e13.
Miller JS, Soignier Y, Panoskaltsis-Mortari A, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105(8):3051–3057.
Romee R, Rosario M, Berrien-Elliott MM, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016;8(357):357ra123.
Shimasaki N, Jain A, Campana D. NK cell-based immunotherapies for hematological malignancies. Blood. 2020;136(5):503–514.
Bachanova V, Cooley S, DeFor TE, et al. Clearance of acute myeloid leukemia by haploidentical NK cells is improved using IL-2 diphtheria toxin fusion protein. Blood. 2014;123(25):3855–3863.
Sun JC, Lanier LL. NK cell development, homeostasis and function: parallels with CD8+ T cells. Nat Rev Immunol. 2011;11(10):645–657.
Screpanti V, Wallin RP, Ljunggren HG, Grandien A. A central role for death receptor-mediated apoptosis in the rejection of tumors by NK cells. J Immunol. 2001;167(4):2068–2073.
López-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of metastasis by NK cells. Cancer Cell. 2017;32(2):135–154.
Lapteva N, Szmania SM, van Rhee F, Rooney CM. Clinical grade purification and expansion of natural killer cells. Crit Rev Oncol Hematol. 2014;91(2):175–186.
Kärre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature. 1986;319(6055):675–678.
Spanholtz J, Tordoir M, Eissens D, et al. High log-scale expansion of functional human natural killer cells from umbilical cord blood CD34+ cells for adoptive cancer immunotherapy. PLoS One. 2010;5(2):e9221.
Miller JS, Morishima C, McNeel DG, et al. A first-in-human phase I study of subcutaneously administered interleukin-15 (rhIL-15) in patients with advanced solid tumors. Clin Cancer Res. 2018;24(7):1525–1535.
Granzin M, Wagner J, Köhl U, et al. Shaping of natural killer cell antitumor activity by ex vivo cultivation. Front Immunol. 2017;8:458.
Denman CJ, Senyukov VV, Gill RG, et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS One. 2012;7(1):e30264.
Klingemann H, Boissel L, Toneguzzo F. Natural killer cells for immunotherapy – advantages of the NK-92 cell line over blood NK cells. Front Immunol. 2016;7:91.
Knorr DA, Bachanova V, Verneris MR, Miller JS, Kaufman DS. Clinical utility of natural killer cells in cancer therapy and transplantation. Semin Immunol. 2014;26(2):161–172.
Orange JS. Formation and function of the lytic NK-cell immunological synapse. Nat Rev Immunol. 2008;8(9):713–725.
Vivier E, Raulet DH, Moretta A, et al. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331(6013):44–49.
Walzer T, Dalod M, Vivier E, Zitvogel L. Natural killer cell–dendritic cell crosstalk in the initiation of immune responses. Expert Opin Biol Ther. 2005;5(Suppl 1):S49–S59.
Cerwenka A, Lanier LL. Natural killer cell memory in infection, inflammation and cancer. Nat Rev Immunol. 2016;16(2):112–123.
Basu S, Binder RJ, Ramalingam T, Srivastava PK. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity. 2001;14(3):303–313.
Shah NN, Baird K, Delbrook C, et al. CD19-targeted CAR NK cells in relapsed or refractory B-cell malignancies: a single-center Phase I clinical trial. J Clin Oncol. 2023;41(2_suppl):11.
Liu E, Tong Y, Dotti G, et al. Cord blood NK cells engineered with IL-15 and a CD19-targeting CAR show long-term persistence and potent antitumor activity. Sci Transl Med. 2020;12(568):eaaz6108.