

Understanding Protein-Based Neurotherapeutics
Protein-based therapeutics are emerging as a critical modality in the treatment of central nervous system (CNS) disorders, offering precise functional modulation at the protein level

The therapeutic landscape for central nervous system (CNS) disorders is undergoing a paradigm shift driven by the advancement of protein and peptide-based biologics. Unlike traditional small molecules, which typically modulate enzymatic activity or receptor signaling from the extracellular space or through orthosteric binding, protein therapeutics offer direct and multifaceted engagement with disease-relevant targets at the proteomic level, the functional layer at which cellular homeostasis, signaling, and structural integrity are executed. These agents include recombinant enzymes, neurotrophic factors, monoclonal antibodies, fusion proteins, and engineered peptides, each designed to intervene at distinct points along the molecular pathophysiology of CNS disease. The inherent specificity, high binding affinity, and biologically encoded functionality of protein therapeutics make them uniquely suited for targeting complex, spatially restricted, or intracellularly located components of neural circuitry.
Historically, the application of protein-based therapies in neurology has been hindered by formidable delivery barriers, most notably the blood-brain barrier (BBB), as well as concerns regarding protein instability, rapid proteolytic degradation, and immunogenicity. However, recent advances in protein engineering, glyco-optimization, and targeted delivery vectors have fundamentally altered the feasibility of protein biologics in CNS applications. Innovative strategies such as receptor-mediated transcytosis, Fc-fusion for neonatal Fc receptor (FcRn) recycling, BBB-penetrant bispecific antibodies, and intrathecal administration platforms have enabled pharmacologically active concentrations of therapeutic proteins to be achieved within neural tissues. Concurrently, the integration of structural biology, computational protein design, and next-generation expression systems has expanded the repertoire of optimizable parameters including half-life, receptor selectivity, folding kinetics, and aggregation resistance, making CNS-targeted protein therapeutics increasingly viable from a manufacturing and clinical standpoint.
The breadth of therapeutic targets accessible via protein modalities is particularly relevant in the context of CNS disease, where multiple levels of dysfunction ranging from genetic enzyme deficiencies and proteotoxic stress to neuroinflammation and circuit-level disintegration may co-exist within a single pathology. Protein therapeutics can provide catalytic activity, as in enzyme replacement therapies, trophic support via neurotrophic factors, immunological modulation through monoclonal antibodies, or high-precision receptor agonism and antagonism via engineered peptides. Furthermore, their modular design enables combination with emerging RNA and gene-based therapies to synergistically modulate both upstream genetic and downstream proteomic determinants of disease. As the field transitions toward personalized and mechanistically targeted treatments, protein-based neurotherapeutics stand poised to become foundational components of next-generation strategies for treating both rare monogenic and common multifactorial CNS disorders.
Protein-based therapeutics are emerging as a critical modality in the treatment of central nervous system (CNS) disorders, offering precise functional modulation at the protein level, which represents the primary effector layer of cellular biology. These agents include recombinant proteins, therapeutic peptides, monoclonal antibodies, fusion proteins, and enzyme replacement therapies. Each is designed to engage endogenous pathways, compensate for molecular deficiencies, or neutralize pathological entities. While RNA-based therapies modulate gene expression before translation, protein therapeutics exert their effects directly at the proteomic interface, where cellular decisions are executed. In the CNS, where spatial and temporal signaling precision is essential, protein-based interventions can act rapidly and with high target selectivity.
Historically, the clinical development of protein and peptide therapeutics for neurological disease has been constrained by the blood-brain barrier (BBB), proteolytic degradation, and immunogenicity. However, recent innovations in protein engineering, delivery vectors, BBB transcytosis strategies, and intrathecal administration have significantly expanded the feasibility and scope of these biologics in neurology. With advances in rational design and molecular targeting, protein-based neurotherapeutics are now addressing a wide range of diseases, from rare genetic enzyme deficiencies to complex, multifactorial neurodegenerative conditions.
Classes and Mechanisms of Action
Neurotrophic Factors and Growth Proteins
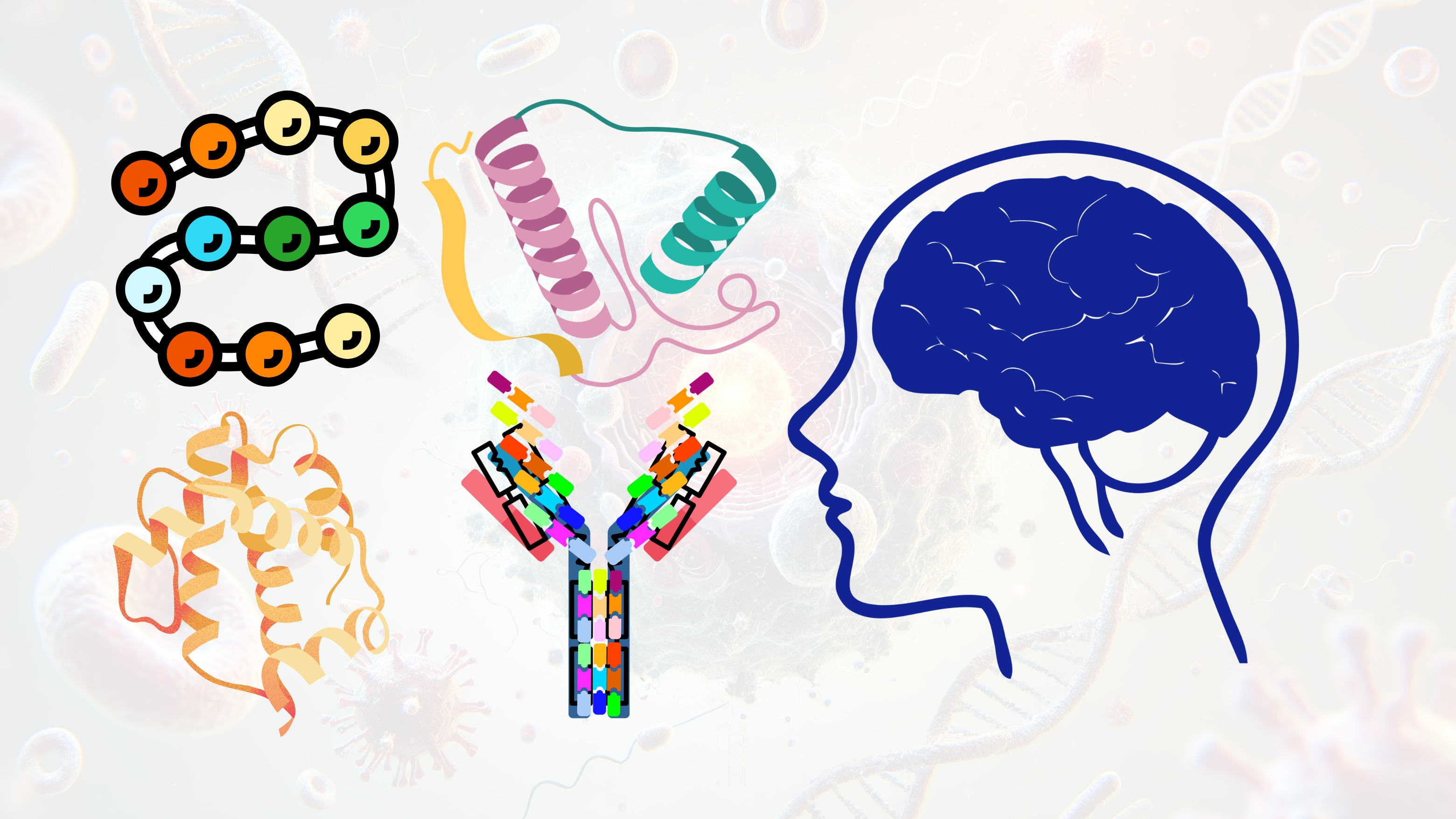
Neurotrophic factors are a class of secreted, protein-based growth regulators that support the survival, development, and functional maintenance of neurons and glial cells in the central nervous system (CNS). These proteins play essential roles in synaptic plasticity, axonal pathfinding, dendritic arborization, and activity-dependent neuronal remodeling. The major neurotrophin family includes brain-derived neurotrophic factor (BDNF), nerve growth factor (NGF), neurotrophin-3 (NT-3), and neurotrophin-4 (NT-4), while other structurally distinct neurotrophic molecules include glial cell line-derived neurotrophic factor (GDNF), ciliary neurotrophic factor (CNTF), and insulin-like growth factor 1 (IGF-1). These proteins signal through distinct but overlapping families of high-affinity receptors, activating intracellular cascades that regulate neuronal survival, metabolism, and gene expression.
Receptor Binding and Signaling Pathways
Classical neurotrophins (NGF, BDNF, NT-3, NT-4) exert their biological effects primarily through binding to the tropomyosin receptor kinase (Trk) family of receptor tyrosine kinases. NGF binds TrkA, BDNF and NT-4 bind TrkB, and NT-3 preferentially binds TrkC, although cross-reactivity exists. Ligand binding induces receptor dimerization and autophosphorylation of tyrosine residues in the intracellular domain, which serves as a docking platform for adaptor proteins such as Shc, FRS2, and IRS-1. These adaptors initiate canonical signaling cascades including the Ras-Raf-MEK-ERK pathway (mitogenic signaling), PI3K-Akt pathway (anti-apoptotic signaling), and PLCγ1 pathway (regulation of intracellular calcium and PKC activation). Each of these pathways converges on transcriptional regulators such as CREB, NF-κB, and FOXO proteins that orchestrate long-term changes in neuronal phenotype.
In addition to Trk receptors, all neurotrophins also bind to the low-affinity p75 neurotrophin receptor (p75^NTR^), a member of the TNF receptor superfamily. p75^NTR^ can modulate Trk receptor affinity and, in the absence of Trk co-expression, can initiate JNK-dependent apoptotic cascades. The dual signaling capacity of neurotrophins via Trk and p75^NTR^ allows for context-dependent effects that vary by cell type, developmental stage, and receptor expression profile.
GDNF family ligands (GDNF, neurturin, artemin, persephin) signal through a two-component system consisting of the GDNF family receptor alpha (GFRα1–4) co-receptors and the transmembrane RET tyrosine kinase. Ligand binding to GFRα induces RET dimerization and phosphorylation, leading to downstream activation of PI3K-Akt, MAPK, and JAK-STAT pathways. RET signaling is particularly critical in dopaminergic neuron maintenance and has been a key target for Parkinson’s disease therapeutics.
CNTF, a member of the IL-6 cytokine family, signals through a tripartite receptor complex composed of CNTFRα, gp130, and LIFRβ. This activates the JAK-STAT pathway, promoting neuroprotection and gliogenesis. IGF-1 acts via the IGF1 receptor, a receptor tyrosine kinase with high structural homology to the insulin receptor. Upon ligand binding, autophosphorylation of IGF1R activates IRS proteins, triggering the PI3K-Akt-mTOR and MAPK cascades. IGF-1 is known to enhance synaptogenesis, dendritic complexity, and plasticity in both developing and adult CNS tissue.
Protein Engineering and Optimization
Native neurotrophic factors suffer from poor pharmacokinetic profiles, limited diffusion in CNS parenchyma, and potential pleiotropic or off-target effects due to broad receptor expression. To address these issues, engineering strategies have focused on increasing receptor specificity, prolonging half-life, and improving brain penetration. This includes:
Point mutations in ligand-binding domains to enhance Trk specificity while reducing p75^NTR^ interaction, minimizing pro-apoptotic signaling.
Fc-fusion proteins, which link neurotrophic factors to IgG Fc domains to leverage FcRn-mediated recycling and extend systemic half-life.
Pegylation or glycosylation modifications to improve solubility, reduce immunogenicity, and prevent rapid renal clearance.
Encapsulation in nanoparticles, liposomes, or hydrogels for controlled release and enhanced brain distribution.
Delivery systems are often designed for either intraparenchymal administration (e.g., convection-enhanced delivery of GDNF) or for systemic delivery with BBB-targeting elements. For example, TrkB agonist antibodies or BDNF mimetic peptides conjugated to transferrin receptor-binding domains are being explored to achieve BBB transcytosis.
The therapeutic utility of neurotrophic factors such as BDNF, GDNF, NGF, and IGF-1 is significantly limited by their poor pharmacokinetic profiles, structural instability, receptor promiscuity, and challenges associated with CNS delivery. To overcome these obstacles, extensive molecular engineering is applied to optimize their primary sequence, structural conformation, post-translational modifications, and systemic behavior. At the sequence level, site-directed mutagenesis is used to enhance receptor selectivity and binding kinetics. For example, BDNF variants with mutations such as R125A or Y103A retain TrkB affinity while significantly reducing binding to p75^NTR^, thereby minimizing pro-apoptotic signaling. In parallel, engineered GDNF mutants alter interaction sites on the GFRα1-RET complex to reduce off-target effects or modulate intracellular signaling bias. Computational design techniques, including Rosetta and molecular dynamics simulations, are used to refine ligand-receptor interfaces based on predicted binding free energy and conformational flexibility.
Post-translational modification is another critical area of optimization. N-linked glycosylation sites are introduced into surface-exposed loops to enhance serum half-life, reduce proteolytic degradation, and limit hepatic clearance. These glycosylation motifs (typically N-X-S/T) are engineered into proteins expressed in mammalian systems to enable proper folding and complex glycan processing, including sialylation for stability and reduced immunogenicity. For example, GDNF variants produced in CHO cells are glycoengineered to favor complex, branched glycans over high-mannose forms. Disulfide bond engineering is also employed to stabilize protein structure by reducing entropy in the unfolded state. New cysteine pairs are introduced based on loop proximity and modeled disulfide bond geometry to reinforce native tertiary structure and prevent aggregation or misfolding, particularly in cystine-knot neurotrophins like NGF and NT-3.
To further increase thermodynamic and kinetic stability, the hydrophobic core is repacked via rational mutagenesis. Substitutions of smaller hydrophobic residues with bulkier ones (e.g., Ala to Leu or Val to Ile) fill voids and improve van der Waals packing interactions, thereby raising the melting temperature (Tm) and ΔG of folding. Simultaneously, electrostatic optimization is achieved by engineering salt bridges or hydrogen bonds across domain interfaces. Charge balance is improved by eliminating buried ionizable residues or introducing stabilizing pairs such as Asp-Arg or Glu-Lys. Aggregation-prone regions are identified using in silico tools such as TANGO, AGGRESCAN, and Zyggregator, and mutated to incorporate polar or charged amino acids that disrupt β-sheet stacking or hydrophobic patching. PEGylation and fusion to sterically bulky domains further mitigate aggregation in both formulation and in vivo settings.
Pharmacokinetic enhancement is central to enabling therapeutic efficacy in systemic or CNS delivery. Fc fusion proteins, in which the neurotrophic factor is genetically fused to the Fc domain of human IgG1, utilize neonatal Fc receptor (FcRn)-mediated recycling to prolong circulatory half-life and protect against renal clearance. These fusions are often engineered with flexible glycine-serine-rich linkers to prevent steric hindrance at the receptor-binding interface. Effector function can be minimized through Fc mutations such as LALA or N297A to avoid complement activation and antibody-dependent cellular cytotoxicity. PEGylation is also used to increase hydrodynamic size and reduce protease susceptibility. Site-specific PEGylation, achieved via engineered cysteines or non-canonical amino acid incorporation, allows precise spatial control to avoid interfering with receptor engagement. GlycoPEGylation, wherein PEG is conjugated to engineered glycans, maintains native protein structure and bioactivity while providing metabolic stability. Lipidation with palmitate or stearate moieties enables reversible binding to serum albumin, thereby creating a depot effect and reducing renal clearance. Alternatively, fusion to albumin-binding domains (ABDs) or nanobody tags achieves similar effects without direct lipid attachment.
Expression system selection also plays a crucial role in the successful production of engineered neurotrophic proteins. Mammalian cell lines such as HEK293 and CHO are favored for their ability to carry out human-compatible glycosylation, disulfide formation, and protein folding. In cases where rapid screening is required, yeast (e.g., Pichia pastoris) or insect cell systems can be employed with codon optimization and protease-deficient strains, although post-translational fidelity may be limited. Emerging approaches in cell-free protein synthesis (CFPS) allow incorporation of non-canonical amino acids, click-chemistry functional groups, or chemically reactive handles for downstream conjugation, offering new routes for modular engineering of neurotrophic therapeutics.
Collectively, these engineering strategies allow for the precise control of neurotrophic protein pharmacodynamics and pharmacokinetics, enabling safer and more effective CNS-targeted therapies. By integrating protein chemistry, structural biology, and recombinant expression technologies, it is now possible to systematically improve native neurotrophins for clinical application, enhancing their translational potential in neurodegenerative, neurodevelopmental, and traumatic CNS disorders.
Clinical and Preclinical Applications
In Parkinson’s disease, GDNF and neurturin have shown the ability to protect and regenerate dopaminergic neurons in the substantia nigra and striatum in animal models. However, clinical trials using intraputaminal infusion have yielded mixed results, likely due to delivery heterogeneity and insufficient coverage of target areas. Engineering more diffusible variants or developing systemically administered derivatives remains a key goal.
BDNF and TrkB agonists are under investigation for Alzheimer’s disease, Huntington’s disease, major depressive disorder, and traumatic brain injury due to their capacity to support synaptic maintenance and reduce neuroinflammation. IGF-1 has shown promise in ALS and Rett syndrome models, where it promotes neuromuscular junction stabilization and dendritic growth. CNTF has been explored in optic neuropathies and spinal cord injury due to its glial trophic effects and anti-apoptotic properties.
Several mimetics and small molecule Trk agonists are also in development, aiming to replicate the neurotrophic effect without the limitations of protein therapeutics. These include 7,8-dihydroxyflavone (a TrkB agonist) and small cyclic peptides that bind selectively to neurotrophin receptors.
Enzyme Replacement Therapies
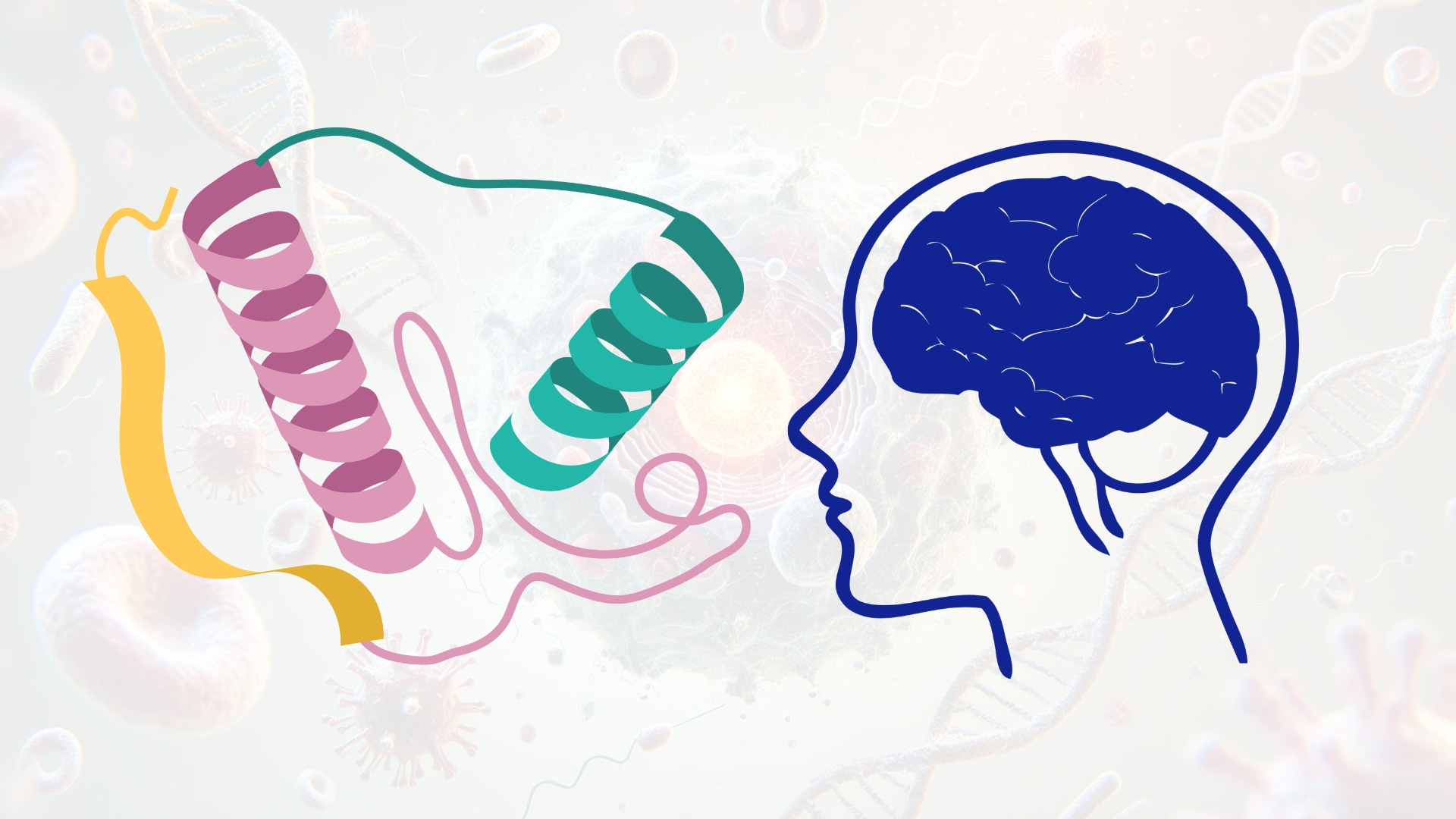
Enzyme deficiencies in the CNS, particularly lysosomal storage disorders, can be addressed by delivering recombinant enzymes that restore catabolic function. Cerliponase alfa, a recombinant form of TPP1, has been approved for CLN2 disease and is administered intraventricularly to bypass the BBB and compensate for absent lysosomal tripeptidyl peptidase 1. Other targets include arylsulfatase A in metachromatic leukodystrophy and β-glucuronidase in mucopolysaccharidosis VII. These proteins are often glycosylated to allow mannose-6-phosphate receptor mediated uptake and lysosomal trafficking. Engineering proteins for CNS delivery via receptor-mediated transcytosis, for example through transferrin or insulin receptor targeting motifs, is extending the reach of enzyme replacement therapies beyond direct CNS infusion.
Enzyme replacement therapies (ERTs) aim to restore deficient or absent lysosomal or metabolic enzyme activity in genetic disorders, particularly lysosomal storage diseases (LSDs). These conditions frequently affect the CNS, where substrate accumulation leads to neurodegeneration, demyelination, and gliosis. ERT involves systemic or local delivery of a recombinant version of the defective enzyme, designed to be internalized by cells and trafficked to the appropriate subcellular compartment, typically the lysosome. To function effectively, therapeutic enzymes must be biochemically active, structurally stable, glycosylated for uptake, and appropriately targeted to the CNS. Achieving this requires precise molecular engineering at multiple levels: primary sequence optimization, post-translational processing, receptor-targeted uptake strategies, and delivery system integration.
Enzyme Structure and Catalytic Activity
Most therapeutic enzymes used in CNS-targeted ERTs are soluble hydrolases that catalyze the degradation of glycosaminoglycans, glycolipids, or nucleic acids within lysosomes. These include β-glucuronidase (MPS VII), tripeptidyl peptidase 1 (CLN2), arylsulfatase A (MLD), α-L-iduronidase (MPS I), and β-galactocerebrosidase (Krabbe disease). Structurally, these enzymes are globular proteins typically composed of α/β hydrolase folds or TIM barrel domains that position catalytic residues in a solvent-accessible active site pocket.
Mutagenesis is frequently applied to improve folding, solubility, or substrate specificity. For example, enhancing catalytic efficiency (k_cat/K_M) can allow therapeutic benefit at lower doses, which is particularly important for CNS applications where enzyme diffusion is limited. Engineering may also reduce off-target activity by narrowing substrate range via loop remodeling or introduction of second-shell mutations that influence active site dynamics. Crystal structures and substrate analog co-crystals are used to guide rational design, supported by molecular dynamics simulations and QM/MM modeling of reaction transition states.
Lysosomal Targeting: Mannose-6-Phosphate Receptor Pathway
For ERT to be effective, the exogenous enzyme must be efficiently taken up by target cells and delivered to the lysosome. This is primarily achieved through the cation-independent mannose-6-phosphate receptor (CI-MPR) pathway. The CI-MPR recognizes N-linked glycans bearing mannose-6-phosphate (M6P) residues on high-mannose oligosaccharide branches. Once bound, the receptor-enzyme complex undergoes clathrin-mediated endocytosis, traffics through early and late endosomes, and is sorted into lysosomes, where the enzyme is released under acidic pH conditions.
Therapeutic enzymes are thus expressed in mammalian systems (e.g., CHO or HEK293 cells) with glycosylation machinery capable of generating M6P-tagged glycans. The efficiency of M6P tagging depends on both the number and accessibility of N-glycosylation sites, and the activity of endogenous GlcNAc-1-phosphotransferase (GNPTAB) and uncovering enzyme (NAGPA). Engineering strategies may include:
Insertion of additional N-X-S/T motifs at solvent-exposed, flexible loops to increase glycan occupancy.
Directed evolution or saturation mutagenesis to identify variants with enhanced M6P modification efficiency.
Expression in engineered cell lines overexpressing GNPTAB and NAGPA or with suppressed competing glycosidases.
Analytical methods such as MALDI-TOF glycoprofiling, HPAEC-PAD, and M6P-specific antibody ELISAs are used to quantify glycan composition and M6P content on therapeutic enzymes.
Alternative Receptor-Targeting Strategies
For CNS delivery, reliance solely on M6P-mediated uptake is often insufficient due to limited access across the BBB and low expression of CI-MPR on neurons. To circumvent this, alternative receptor-targeting domains are genetically fused to the enzyme:
Transferrin receptor (TfR) ligands (e.g., monoclonal antibodies or peptide mimetics) enable receptor-mediated transcytosis across the BBB. Enzyme-TfR fusions bind to TfR on brain endothelial cells and are transported via vesicular pathways into the CNS parenchyma.
Insulin receptor (INSR) targeting peptides and low-density lipoprotein receptor-related protein 1 (LRP1) ligands are also used to exploit endogenous transport systems.
Fusion to ApoE-derived peptides, albumin-binding domains, or cell-penetrating peptides (e.g., TAT) enhances uptake into neurons and glia independent of M6P.
Importantly, linker length, orientation, and valency of the targeting moiety must be carefully optimized to preserve both receptor binding and catalytic activity. Flexible (Gly₄Ser)n linkers are commonly used to spatially decouple domains and maintain independent folding. Biophysical methods such as surface plasmon resonance (SPR) and isothermal titration calorimetry (ITC) quantify receptor binding kinetics (K_D, k_on, k_off), while enzyme assays confirm retained activity post-fusion.
Protein Stability and Aggregation Resistance
Therapeutic enzymes must resist denaturation, aggregation, and proteolysis during formulation, circulation, and intracellular trafficking. Key strategies include:
Disulfide engineering: New disulfide bonds are introduced to reduce entropy of the unfolded state and improve thermal stability. These are modeled using distance constraints (Cβ–Cβ ~ 4.5–6.5 Å) and validated via differential scanning calorimetry (DSC).
Charge optimization: Surface electrostatics are modified to reduce aggregation by eliminating patches of net positive or negative charge that promote non-specific interactions. pI tuning and buried salt bridge formation can also increase pH stability.
Deamidation and oxidation resistance: Substitution of labile residues (e.g., Asn-Gly, Met) in solvent-exposed regions minimizes chemical degradation during storage or in vivo.
PEGylation: Covalent attachment of polyethylene glycol (PEG) chains masks protease cleavage sites, increases hydrodynamic radius, and prevents aggregation. Site-specific PEGylation is often performed at engineered cysteines or non-canonical amino acids (e.g., azidohomoalanine) introduced by genetic code expansion.
CNS Delivery: Routes and Vehicles
The most direct route for CNS-targeted ERT is intraventricular or intrathecal injection, which bypasses the BBB and delivers the enzyme directly into cerebrospinal fluid (CSF). Once in the CSF, enzymes must distribute through perivascular spaces, penetrate brain parenchyma, and be taken up by target cells. Parenchymal diffusion is limited by molecular size, charge, and extracellular matrix binding. Engineering enzymes with reduced isoelectric point (pI) or surface charge neutrality can enhance interstitial mobility.
For systemic administration, delivery vehicles are used to protect enzymes and facilitate CNS access:
Lipid nanoparticles (LNPs) and polymeric nanoparticles (e.g., PLGA) can encapsulate enzymes, shield them from degradation, and incorporate BBB-targeting ligands.
Exosomes, derived from engineered donor cells, naturally cross the BBB and can carry lysosomal enzymes either encapsulated or surface-bound.
AAV-based gene therapy can deliver cDNA encoding the enzyme to CNS cells, but in hybrid ERT strategies, short-term protein supplementation is used to accelerate therapeutic onset before gene expression reaches steady-state levels.
Manufacturing and Biochemical Characterization
Recombinant enzymes for ERT are produced in GMP-grade mammalian systems, typically CHO or HEK293 cells, to ensure proper folding and glycosylation. Purification includes affinity chromatography (e.g., M6P affinity resins), ion-exchange, and SEC to remove aggregates. Key analytical assays include:
Enzyme activity assays using fluorogenic or chromogenic substrates to measure k_cat and K_M.
Glycan profiling via LC-MS, HPAEC, or lectin blotting to quantify M6P content.
Structural confirmation by circular dichroism (CD), thermal shift assays, or limited proteolysis.
Receptor binding assays (e.g., SPR) to confirm ligand-target engagement.
Uptake and trafficking via confocal microscopy, lysosomal co-localization (e.g., LAMP1 staining), and subcellular fractionation in relevant cell models.
In summary, enzyme replacement therapies for CNS applications require an integrated approach that combines precise enzymology, glycoengineering, receptor targeting, structural stabilization, and delivery optimization. The molecular biology and biochemistry underlying these interventions dictate not only the catalytic competence of the enzyme, but also its biodistribution, cellular uptake, and long-term efficacy in neurodegenerative lysosomal disorders. Ongoing innovations in protein design, receptor biology, and CNS drug delivery are continuously enhancing the therapeutic index of ERTs, bringing them closer to broader application in neurological medicine.
Monoclonal Antibodies
Monoclonal antibodies (mAbs) have shown utility in CNS immunomodulation and protein aggregate clearance. Antibodies targeting amyloid-beta, such as aducanumab, tau, alpha-synuclein, or TDP-43 are under investigation or approved for Alzheimer’s and Parkinson’s disease. These mAbs can neutralize extracellular aggregates, promote microglial phagocytosis via Fc receptor engagement, or prevent intercellular propagation of pathogenic seeds. Additional applications include checkpoint inhibitors in glioblastoma and antibodies against complement proteins or cytokines in neuroinflammatory conditions. Engineering of antibody Fc regions, valency, and glycosylation profiles is used to tune effector function, half-life, and BBB penetration.
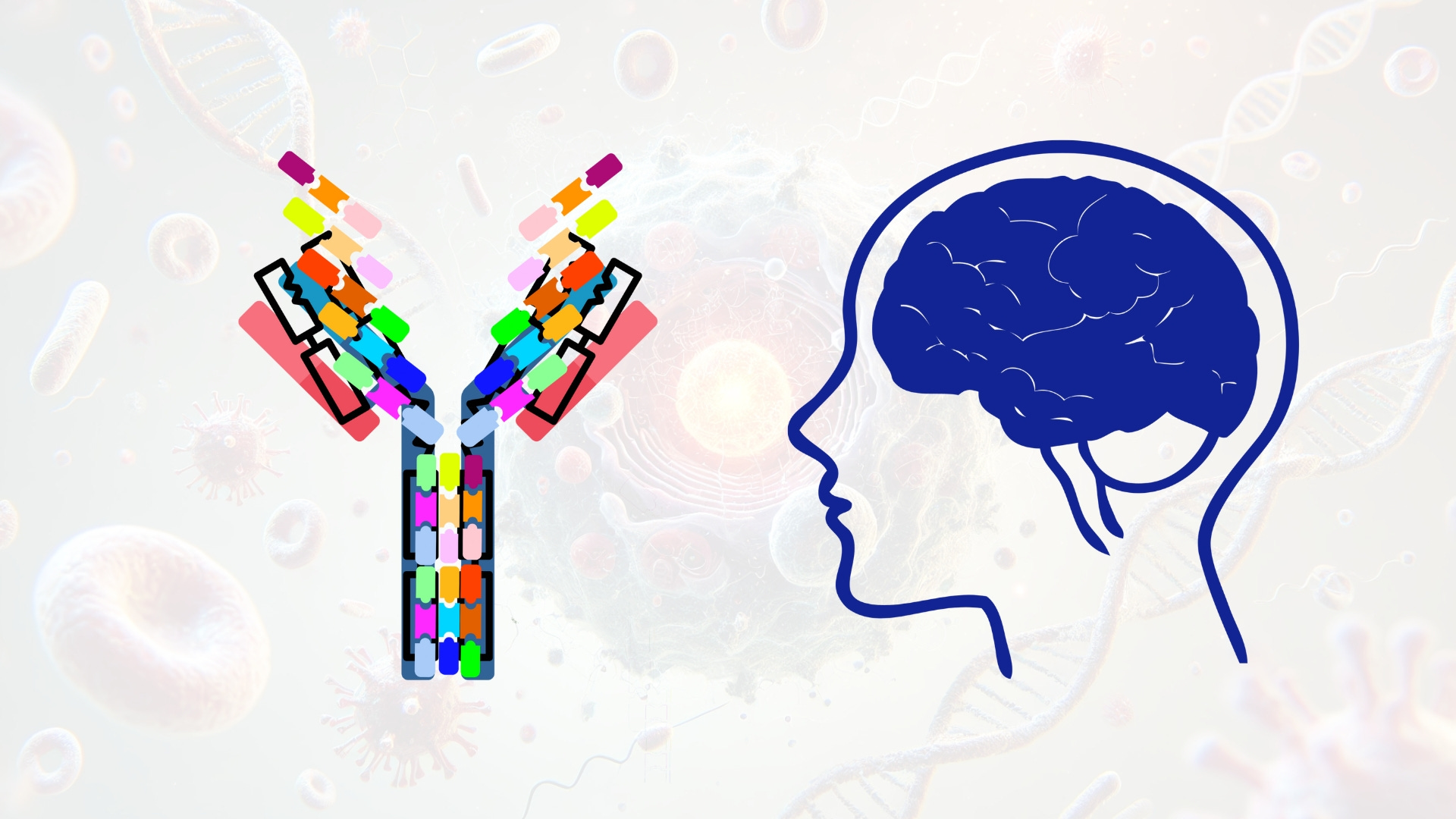
Monoclonal antibodies (mAbs) are highly specific immunoglobulin-based therapeutics that exert their activity through high-affinity antigen recognition and, in some cases, engagement of immune effector functions. Structurally, mAbs consist of two identical heavy chains and two identical light chains, joined by disulfide bonds to form a Y-shaped molecule with a molecular weight of approximately 150 kDa. The variable regions of the heavy and light chains (VH and VL) form the antigen-binding fragment (Fab), where six complementarity-determining regions (CDRs) create the paratope. The Fc (fragment crystallizable) region is composed of the CH2 and CH3 domains of the heavy chains and is responsible for interactions with Fc gamma receptors (FcγRs), complement proteins (such as C1q), and the neonatal Fc receptor (FcRn), which recycles IgG and extends its serum half-life.
In CNS-targeted therapeutics, mAbs are developed to neutralize or clear pathological proteins such as amyloid-β (Aβ), hyperphosphorylated tau, α-synuclein, and TDP-43, or to modulate immune signaling pathways, such as PD-1/PD-L1 in glioblastoma. To achieve this, the variable regions of mAbs are engineered for high affinity and specificity to disease-associated epitopes. Phage display, deep mutational scanning, and next-generation sequencing-guided selection are used to evolve antibodies with optimized CDR sequences. Structural characterization of antigen-antibody complexes by X-ray crystallography or cryo-electron microscopy enables identification of conformational epitopes and guides affinity maturation. For aggregation-specific targeting, antibodies are designed to selectively recognize misfolded or oligomeric forms of proteins while sparing physiological monomers, reducing the risk of disrupting normal protein function.
The Fc region is engineered to control effector function, pharmacokinetics, and biodistribution. For most CNS applications, where inflammation is detrimental, effector function is minimized. Point mutations such as L234A/L235A (LALA), N297A, or P329G disrupt FcγR and complement C1q binding, thereby abolishing antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). To enhance serum half-life, mutations like M428L/N434S (LS) or the triple mutant M252Y/S254T/T256E (YTE) increase affinity for FcRn at acidic pH, promoting recycling and prolonged systemic exposure. Additionally, glycoengineering is used to modulate Fc function. For example, reducing core fucosylation can enhance ADCC, while sialylation can suppress inflammatory responses. For CNS use, maintaining a fully fucosylated, non-inflammatory glycan profile is generally preferred.
The major pharmacological barrier for mAbs in neurology is the blood-brain barrier (BBB), which prevents nearly all large proteins from entering the CNS by passive diffusion. Under normal conditions, less than 0.2% of a systemically administered IgG reaches brain parenchyma. To address this, antibodies are engineered for receptor-mediated transcytosis (RMT) using bispecific designs. These molecules contain one Fab arm targeting the therapeutic antigen and another binding to a BBB-expressed receptor, such as the transferrin receptor (TfR) or insulin receptor (INSR). Monovalent binding to TfR with moderate affinity is critical to prevent lysosomal sequestration and enable transcytosis across endothelial cells. The use of anti-TfR single-chain Fvs or peptides fused to the Fc or Fab regions has been successful in preclinical models for increasing CNS uptake. Alternative strategies include nanobodies and single-domain antibodies (V_HH), which are smaller (~15 kDa), more stable, and capable of deeper tissue penetration. These formats can be conjugated to neuroactive payloads or expressed in the CNS using adeno-associated virus (AAV) vectors for chronic conditions.
Stability and solubility are critical for CNS-deployable mAbs. Engineering the framework regions of the VH and VL domains helps raise thermal melting temperature (T_m), reduce hydrophobic aggregation hotspots, and eliminate post-translational liabilities such as deamidation-prone Asn-Gly sequences or oxidation-sensitive Met residues. Antibodies are formulated in optimized buffers to preserve structure during storage and delivery, with attention to pH, ionic strength, and excipients such as polysorbates or trehalose. Biophysical characterization includes differential scanning calorimetry (DSC) for thermal stability, size exclusion chromatography (SEC) for aggregate content, and dynamic light scattering (DLS) for colloidal stability.
Manufacturing is typically performed in CHO cell lines optimized for high yield and human-compatible glycosylation. The resulting antibodies are purified through Protein A affinity chromatography, followed by ion exchange and polishing steps. Critical quality attributes include monomer content, glycan profile, charge heterogeneity, and endotoxin level, all of which are essential for clinical-grade production. In vitro potency assays, such as antigen-binding ELISA, receptor-blocking assays, or cell-based activity readouts, are used to confirm functionality, along with binding kinetics analysis by surface plasmon resonance (SPR) or biolayer interferometry (BLI).
Several mAbs have advanced to clinical use or late-stage trials in neurology. Aducanumab and lecanemab are approved for Alzheimer’s disease and target aggregated Aβ with different conformational preferences. Anti-tau antibodies, such as tilavonemab and semorinemab, are under development for progressive supranuclear palsy and Alzheimer’s. Anti-α-synuclein antibodies have been explored for Parkinson’s disease, while anti-TDP-43 and anti-SOD1 antibodies are being tested in ALS. Immune checkpoint inhibitors, such as anti-PD-1 antibodies, are also being trialed in glioblastoma, aiming to reactivate tumor-infiltrating lymphocytes within the CNS microenvironment.
Overall, monoclonal antibody therapeutics for CNS disorders require precise molecular engineering to overcome systemic and neural delivery barriers, modulate immunogenicity, and retain high target affinity in the unique milieu of the brain. Advances in bispecific architecture, receptor targeting, Fc silencing, and antibody stability are expanding the feasibility of antibody-based treatment for neurological diseases. Future progress will likely involve integration with RNA or gene-based therapies, allowing synergistic modulation of both protein expression and function in the CNS.
Therapeutic Peptides
Peptides offer a compact and customizable modality for modulating receptors, ion channels, and protein-protein interactions in the CNS. Examples include conotoxins for voltage-gated calcium channel inhibition, opioid receptor agonist peptides, and NMDA receptor antagonists for depression or excitotoxicity. Therapeutic peptides can mimic endogenous ligands or act as decoys to block pathological signaling. Peptide optimization strategies include cyclization, incorporation of D-amino acids, lipidation, and PEGylation to enhance proteolytic stability, blood-brain barrier permeability, and receptor affinity.
Therapeutic peptides are short, synthetic or recombinant chains of amino acids—typically ranging from 5 to 50 residues—that function by mimicking or modulating protein-protein interactions, receptor-ligand binding events, ion channel activity, or intracellular signaling cascades. In the central nervous system (CNS), where spatially and temporally precise modulation of signaling is critical, peptides offer distinct pharmacodynamic advantages over small molecules due to their high specificity, reduced off-target effects, and compatibility with endogenous pathways. Their relatively low molecular weight compared to full-length proteins facilitates diffusion in extracellular matrices and, when engineered appropriately, can allow for selective permeability across the blood-brain barrier (BBB) or uptake into neural cells.
Peptides exert therapeutic effects in the CNS through a variety of mechanisms. Some function as receptor agonists or antagonists, binding G protein–coupled receptors (GPCRs), ion channels, or receptor tyrosine kinases (RTKs). Examples include synthetic analogs of neuropeptides such as substance P, neuropeptide Y (NPY), oxytocin, or orexin, which regulate neurotransmitter release, neuroendocrine tone, and behavioral states. Others act as enzyme inhibitors, scaffolding disruptors, or decoy ligands that interfere with pathological protein interactions. For instance, peptides targeting the N-methyl-D-aspartate (NMDA) receptor complex can modulate excitotoxicity, while peptides derived from amyloid-β-binding motifs can inhibit aggregation and seeding in Alzheimer’s disease. Some therapeutic peptides are cell-penetrating and act intracellularly to inhibit kinases, transcription factors, or apoptotic mediators, often through allosteric inhibition or competitive binding to interaction domains such as SH3, PDZ, or WW modules.
At the molecular level, the activity of peptides is highly dependent on their conformational stability, proteolytic resistance, and physicochemical compatibility with the CNS microenvironment. Linear peptides in aqueous solution are intrinsically flexible and often lack a stable secondary structure, making them susceptible to rapid degradation by exopeptidases and endopeptidases such as aminopeptidase N, neprilysin, or prolyl oligopeptidase. To overcome this, extensive chemical modifications are introduced to stabilize conformation and protect against enzymatic cleavage. Cyclization is a widely employed technique, including both head-to-tail (N-to-C) and side-chain-to-side-chain linkages (e.g., disulfide, lactam, or thioether bridges), which reduce conformational entropy and promote α-helical or β-turn secondary structure. Backbone cyclization using peptide isosteres or synthetic spacers can further increase resistance to proteolysis without compromising receptor binding.
Incorporation of D-amino acids (the non-natural enantiomers of L-amino acids) at enzymatically labile positions significantly enhances half-life by resisting recognition by stereospecific proteases. Partial or full retro-inverso designs—where the sequence is reversed and all residues are converted to D-chirality—can preserve receptor binding while conferring extensive stability. N-terminal acetylation, C-terminal amidation, and alpha-methylation of labile residues (e.g., Ser, Thr, Tyr) are also applied to reduce chemical hydrolysis and oxidation. PEGylation of side chains, particularly on lysines or cysteines, increases hydrodynamic radius, reduces renal clearance, and can shield immunogenic epitopes. Other stability enhancements include lipidation (e.g., palmitoylation) to promote albumin binding or depot formation, and glycosylation to influence solubility and bioavailability.
Targeting the CNS with therapeutic peptides presents significant delivery challenges, primarily due to the BBB. However, peptides can be designed or modified for enhanced transport. Some peptides inherently cross the BBB via carrier-mediated transporters, such as the large neutral amino acid transporter (LAT1), or receptor-mediated transcytosis, utilizing ligands for transferrin receptor (TfR), insulin receptor (INSR), or low-density lipoprotein receptor-related proteins (LRP1/2). To exploit these routes, chimeric constructs are developed where a BBB-shuttling domain is fused to the therapeutic peptide either covalently or via a cleavable linker. Cell-penetrating peptides (CPPs), such as TAT (from HIV-1), penetratin (from Antennapedia), or arginine-rich motifs, are frequently fused to therapeutic peptides to facilitate translocation across both the BBB and cell membranes. These often act through direct membrane interaction or endocytosis, followed by endosomal escape.
Peptides can also be encapsulated in nanoparticles, liposomes, or hydrogels to improve pharmacokinetics and enhance delivery to brain parenchyma. For instance, PEGylated liposomes functionalized with transferrin ligands or ApoE mimetics have shown increased delivery to CNS targets following intravenous administration. Intranasal delivery is another non-invasive route under development, relying on olfactory and trigeminal nerve uptake to bypass the BBB entirely. Additionally, intrathecal and intraparenchymal injections are used for peptides that require localized and sustained CNS exposure, particularly in neurooncology and neurodegenerative diseases.
Functionally, peptide therapeutics in the CNS cover a broad range of indications. In Alzheimer’s disease, β-sheet breaker peptides are designed to bind and destabilize amyloid fibrils, while tau aggregation inhibitors target hexapeptide motifs (e.g., PHF6) critical for tau nucleation. In Parkinson’s disease, α-synuclein aggregation blockers and mitochondrial-targeting antioxidant peptides (e.g., SS-31) are under investigation. In stroke and traumatic brain injury, neuroprotective peptides targeting NMDA receptor subunits (e.g., GluN2B) or caspase-activating domains have been shown to reduce excitotoxicity and apoptosis. For chronic pain, conotoxins and other peptide ion channel blockers inhibit calcium or sodium currents involved in nociceptive transmission, with agents like ziconotide (a Cav2.2 blocker) already approved for intrathecal use. In psychiatric disorders, peptide mimetics of oxytocin or vasopressin are in development for treating autism spectrum disorder, anxiety, and depression.
Peptides also serve as ligands for neuromodulatory GPCRs, where biased agonism can be engineered to favor beneficial intracellular signaling pathways. For example, modified opioid peptides can activate G protein signaling while avoiding β-arrestin recruitment, thereby reducing tolerance and respiratory depression. Structure-activity relationship (SAR) studies, often guided by NMR or crystallographic data, are used to design such functionally selective peptides with fine-tuned receptor engagement.
From a biomanufacturing perspective, therapeutic peptides are synthesized using solid-phase peptide synthesis (SPPS), which allows for the incorporation of non-natural amino acids, chemical modifications, and macrocyclization. Purification is achieved using high-performance liquid chromatography (HPLC), with characterization via mass spectrometry, circular dichroism, and in vitro binding or activity assays. Advances in automated synthesis and peptide stapling techniques now enable the production of stable, cell-permeable, helical peptides with drug-like properties, expanding the scope of intracellular targets.
In summary, therapeutic peptides for CNS applications represent a rapidly evolving modality that bridges the specificity of biologics with the synthetic flexibility of small molecules. Their modular design, tunable structure, and ability to engage both extracellular and intracellular targets make them particularly suited for diseases where spatial and temporal precision is required. Overcoming delivery and stability challenges through biochemical and structural optimization is central to their clinical success, and ongoing innovations in peptide chemistry, delivery vehicles, and receptor biology are expanding their therapeutic potential across a wide spectrum of neurological diseases.
Therapeutic peptides are defined by their amino acid composition, sequence, and secondary structure, which collectively govern their biochemical behavior, target engagement, and biological activity. They typically range from 5 to 50 amino acids in length, occupying an intermediate space between small molecules and full-length proteins. Their activity is predicated on mimicking endogenous bioactive peptides, modulating protein-protein interactions, or engaging specific receptor targets, often with high specificity and affinity.
Peptide Structure: Sequence and Conformation
At the primary sequence level, therapeutic peptides are constructed from standard L-α-amino acids, though incorporation of non-canonical residues is common to improve stability and function. Sequence dictates the physicochemical properties such as charge distribution (governed by the pKa values of side chains), hydrophobicity (important for membrane permeability), and isoelectric point (pI), which influence solubility and biodistribution. Secondary structure is critical for bioactivity and is commonly stabilized in therapeutic peptides to preserve functional geometry. α-helices, β-hairpins, β-sheets, and loop motifs are stabilized via cyclization, hydrogen bonding, or incorporation of helix-inducing residues (e.g., Ala, Leu, Glu).
Peptides are often disordered in aqueous solution, necessitating stabilization through conformational constraints. Helical stabilization is achieved via hydrocarbon stapling, where α,α-disubstituted non-natural amino acids are covalently linked through olefin metathesis. β-turns and loops can be enforced using D-Pro-L-Pro dipeptides, or backbone N-methylation, which disrupts hydrogen bonding and enforces torsional restrictions. Cyclization, both head-to-tail and side-chain-to-side-chain (e.g., disulfide, lactam), reduces entropic cost of binding and prevents degradation by exopeptidases. Structural characterization via NMR, circular dichroism (CD), and X-ray crystallography is essential for confirming desired conformations.
Receptor Binding and Signaling Mechanisms
Many therapeutic peptides act as ligands for transmembrane receptors, including G protein–coupled receptors (GPCRs), ion channels, and receptor tyrosine kinases (RTKs). These interactions are governed by shape complementarity, electrostatics, and hydrogen bonding between the peptide and the extracellular domain of the receptor. For GPCRs, peptides often mimic the N-terminal domain of endogenous ligands, occupying the orthosteric binding site and inducing conformational changes that facilitate coupling to intracellular G proteins or β-arrestins.
Receptor binding affinity (K_D), kinetics (k_on and k_off), and efficacy (E_max) are typically quantified using radioligand binding assays, surface plasmon resonance (SPR), or isothermal titration calorimetry (ITC). Peptides can act as full agonists, partial agonists, antagonists, or inverse agonists. In engineered peptides, biased agonism is a key concept where ligand-induced receptor conformations preferentially activate specific signaling pathways—such as G protein over β-arrestin signaling in opioid receptors—to improve therapeutic selectivity and reduce side effects.
For intracellular targets, peptides often inhibit protein-protein interactions by mimicking linear or helical epitopes that occupy critical binding grooves, such as SH2, PDZ, or bromodomains. These interactions are inherently shallow and large-surfaced, making them difficult to drug with small molecules but amenable to high-affinity peptides. Achieving intracellular delivery often requires cell-penetrating peptides (CPPs), such as polyarginine, TAT, or penetratin, which promote endocytosis and endosomal escape.
Proteolytic Stability and Pharmacokinetics
Peptides are highly susceptible to enzymatic degradation by proteases in the gastrointestinal tract, plasma, and intracellular compartments. Enzymes such as trypsin, chymotrypsin, neprilysin, angiotensin-converting enzyme (ACE), and aminopeptidases cleave peptides at specific residues, drastically limiting their half-life. To overcome this, multiple strategies are employed:
D-amino acid substitution at cleavage sites disrupts stereospecific recognition by proteases.
N-methylation of amide bonds reduces hydrogen bonding and sterically hinders peptidase activity.
Peptoid backbones (N-substituted glycines) are non-natural and fully resistant to proteolysis.
Retro-inverso peptides, composed of D-amino acids in reverse order, maintain side-chain topology while resisting enzymatic degradation.
Capping of termini via N-acetylation and C-amidation blocks exopeptidase activity.
Pharmacokinetic properties such as absorption, distribution, metabolism, and excretion (ADME) are heavily influenced by peptide size, polarity, and proteolytic resistance. Conjugation to polyethylene glycol (PEGylation), fatty acids (e.g., palmitoylation, myristoylation), or albumin-binding domains can significantly extend plasma half-life by reducing renal clearance and increasing serum protein binding. PEG chains also provide steric protection against proteases and immune recognition. Lipidation enhances binding to serum albumin and facilitates interaction with cell membranes, improving bioavailability.
Chemical Synthesis and Peptide Engineering
Most therapeutic peptides are synthesized by solid-phase peptide synthesis (SPPS) using Fmoc (9-fluorenylmethoxycarbonyl) chemistry. Automated synthesis platforms allow for rapid assembly of sequences and incorporation of non-natural residues, isosteres, or chemically reactive side chains for conjugation. Cyclization is performed either on-resin or in solution, using orthogonally protected residues to form disulfide bridges (oxidative folding), lactam bonds (amide cyclization), or thioether linkages (maleimide chemistry).
Peptide libraries for screening are generated via combinatorial chemistry, phage display, or mRNA display, enabling the identification of high-affinity ligands or inhibitors against targets that are difficult to address with small molecules. Computational tools (e.g., Rosetta FlexPepDock, PEP-FOLD) are used for structural modeling and prediction of peptide binding conformations and energetics. Post-synthetic modifications, including biotinylation, fluorescent tagging, radiolabeling, or attachment of cleavable linkers, facilitate imaging, target validation, and therapeutic delivery.
Formulation and Stability
Peptides are formulated for various routes of administration including subcutaneous, intravenous, intranasal, transdermal, or intrathecal delivery. Lyophilization and the use of stabilizers such as trehalose, mannitol, and surfactants (e.g., polysorbate 80) are essential for preserving structure and preventing aggregation. Formulations must maintain peptide conformation, prevent chemical degradation (e.g., oxidation of Met, deamidation of Asn), and ensure redissolution kinetics compatible with therapeutic delivery.
Stability testing includes differential scanning calorimetry (DSC), high-performance liquid chromatography (HPLC), mass spectrometry (MS), and accelerated degradation studies under stress conditions. Aggregation propensity is assessed via dynamic light scattering (DLS) and thioflavin T fluorescence if amyloidogenic sequences are involved.
Immunogenicity and Tolerability
Although generally less immunogenic than proteins, peptides can still elicit immune responses if they contain T-cell or B-cell epitopes. Immunogenicity is minimized by:
Epitope mapping and deimmunization, using algorithms like NetMHCIIpan to predict MHC class II binding and eliminate immunodominant sequences.
PEGylation or glycosylation to mask epitopes and reduce immune surveillance.
Use of endogenous sequences or derivatives of human peptides to enhance tolerance.
Preclinical immunogenicity assessment involves in vitro assays using human peripheral blood mononuclear cells (PBMCs) and cytokine release profiling, followed by in vivo animal testing for anti-drug antibody (ADA) development.
In summary, therapeutic peptides are chemically and biologically versatile agents that integrate molecular precision, tunable pharmacology, and modifiable structure into a single therapeutic platform. Their efficacy depends critically on the understanding and manipulation of their primary sequence, conformational preferences, receptor interactions, and metabolic fate. As the biochemical toolkit for peptide engineering continues to expand, including stapling, macrocyclization, and non-canonical chemistry, peptides are poised to address previously intractable targets in both extracellular and intracellular spaces. Their modularity and customizability make them invaluable in modern drug development across diverse therapeutic areas.
Molecular Engineering and Optimization
Structural Stability and Pharmacokinetics
Protein-based therapeutics require stabilization against thermal denaturation and proteolytic cleavage. Strategies include the introduction of disulfide bonds, alpha-helical stabilizing motifs, and fusion to the immunoglobulin Fc domain or human serum albumin. These modifications extend half-life by leveraging neonatal Fc receptor mediated recycling and reduce renal filtration due to increased molecular weight. Glycoengineering is also used to enhance plasma stability, reduce immunogenicity, and improve receptor binding.
Targeting and Specificity
Site-specificity is engineered at the sequence level through rational design, phage display, or computational modeling. For BBB penetration, therapeutic proteins are fused to ligands or antibodies targeting receptors such as transferrin receptor, insulin receptor, or LRP1. Bispecific formats, where one arm engages a BBB receptor and the other engages the CNS target, are under active development. For intracellular delivery, proteins may be conjugated to cell-penetrating peptides or encapsulated in nanoparticles. In the case of peptides, lipidation or glycosylation may be employed to promote neuronal uptake and target localization.
Delivery Challenges and Solutions
Blood-Brain Barrier Penetration
The BBB severely restricts the passive diffusion of proteins due to their size and polarity. Intrathecal and intracerebroventricular routes allow direct administration to the CNS and are used in approved therapies such as cerliponase alfa. For systemic delivery, receptor-mediated transcytosis is the most viable approach. Therapeutics are fused to ligands recognized by endothelial receptors, enabling vesicular transport into brain parenchyma. Novel BBB-shuttling antibodies and engineered transport peptides are expanding the toolbox for brain delivery.
Formulation and Route of Administration
Proteins and peptides are sensitive to aggregation and degradation during formulation. Buffer optimization, lyophilization, and excipient selection are used to maintain structural integrity. Intravenous and subcutaneous administration are common, but for CNS indications, intrathecal delivery remains the gold standard for direct access. Convection-enhanced delivery is also being explored to achieve broad parenchymal distribution through positive-pressure infusion.
Clinical Applications and Emerging Therapies
Approved protein-based therapies for CNS disorders include cerliponase alfa for CLN2 disease, aducanumab for Alzheimer’s disease, and onasemnogene abeparvovec, a gene therapy expressing a therapeutic protein for spinal muscular atrophy. Other candidates in late-stage development include neurotrophic factors for Parkinson’s disease, anti-tau antibodies for Alzheimer’s, and monoclonal antibodies for multiple sclerosis. Peptide-based therapies are advancing for epilepsy, pain, depression, and neuroinflammation.
Therapeutic proteins are also being combined with gene therapy and mRNA delivery platforms. For example, AAV-delivered neurotrophins or synthetic mRNAs encoding enzymes are designed to achieve sustained, localized protein expression in the CNS. These hybrid modalities bridge the distinction between gene-based and protein-based therapy and provide flexibility in dosing and reversibility.
Future Directions and Conclusion
Protein and peptide therapeutics offer a unique capacity for direct modulation of molecular and cellular functions in the CNS, particularly when rapid or reversible intervention is required. The intrinsic specificity of protein-protein and ligand-receptor interactions enables targeted therapeutic effects with minimal off-target engagement. Although delivery remains a principal challenge, advances in transcytosis engineering, molecular stabilization, and direct CNS infusion techniques are expanding the accessibility of these biologics to neural tissues.
As the CNS drug development landscape evolves, protein-based therapeutics are expected to play a central role in the treatment of both rare genetic and complex acquired neurological diseases. The modular nature of these agents, combined with the precision of recombinant engineering, positions them as ideal complements to RNA- and gene-based modalities. In the coming decade, therapeutic proteins are likely to become integral components of multimodal treatment regimens, leveraging the advantages of both extracellular signaling modulation and intracellular enzyme replacement. With continued progress in delivery science and protein engineering, protein-based neurotherapeutics will be a cornerstone of next-generation precision medicine in neurology.
Protein-based therapeutics have emerged as a critical class of interventions for central nervous system disorders, offering mechanistically diverse, highly specific, and functionally versatile treatment modalities. Their capacity to engage disease biology directly at the level of proteins—the primary effectors of cellular signaling, metabolism, and structure—allows for therapeutic strategies that extend beyond the capabilities of small molecules or gene-based interventions alone. Through precise modulation of protein function, degradation, signaling, or replacement, these biologics can address a broad spectrum of CNS pathologies, including neurodegeneration, lysosomal storage disorders, neuroinflammation, and circuit dysfunction.
Recent advances in molecular engineering have significantly expanded the therapeutic potential of proteins in the CNS. Innovations in rational design, sequence optimization, post-translational modification, and fusion architecture have yielded molecules with enhanced stability, half-life, receptor specificity, and reduced immunogenicity. Concurrently, the development of advanced delivery platforms, including receptor-mediated transcytosis, intrathecal infusion, and nanoparticle encapsulation, has overcome many of the historical limitations imposed by the blood-brain barrier. These technologies have enabled protein therapeutics to achieve pharmacologically relevant concentrations in neural tissues, opening new avenues for clinical translation.
Looking ahead, protein-based neurotherapeutics are likely to become increasingly integrated into multimodal treatment paradigms that combine biologics, gene therapy, and RNA-based platforms. Their modularity, reversibility, and capacity for fine-grained biochemical targeting make them ideally suited for personalized medicine approaches in neurology. As delivery technologies continue to mature and the molecular underpinnings of CNS diseases are further elucidated, protein therapeutics will occupy a central role in the design of next-generation interventions aimed at restoring function, halting progression, and ultimately reversing the course of neurological disease.
Will any of this treat the autonomic nervous system?