

Understanding RNA Neurotherapeutics
RNA-based therapeutics are rapidly emerging as a class of medicines for central nervous system (CNS) disorders, leveraging the central dogma of molecular biology to modulate gene expression.


RNA-based therapeutics have emerged as a transformative platform in the treatment of central nervous system (CNS) disorders, offering a level of target specificity and mechanistic versatility that surpasses conventional small molecules and biologics. By directly engaging the post-transcriptional regulation of gene expression, RNA modalities enable precise silencing, splicing correction, transcript editing, or de novo protein synthesis without the need for genomic modification. This class of therapeutics includes antisense oligonucleotides (ASOs), small interfering RNAs (siRNAs), messenger RNAs (mRNAs), microRNA (miRNA) modulators, long non-coding RNA (lncRNA) agents, and site-directed RNA editing systems, each tailored to address distinct molecular defects underlying neurological diseases.
Their modular design and programmable sequence complementarity make them particularly suited for targeting the complex and heterogeneous gene expression landscapes characteristic of CNS pathologies. While delivery across the blood-brain barrier and immunogenicity remain key challenges, recent advancements in chemical modifications, delivery vectors, and intrathecal administration techniques have significantly expanded the clinical viability of RNA therapeutics in neurology. As a result, RNA-based interventions are rapidly advancing from experimental tools to clinically validated treatments, redefining the therapeutic potential for both genetic and acquired CNS disorders.
These therapies intervene at the RNA level to either suppress, replace, correct, or augment protein expression, offering new avenues to treat diseases that have historically been intractable with small molecules or monoclonal antibodies. Recent progress in synthetic chemistry, molecular engineering, and delivery systems has catalyzed the development of RNA therapeutics that are now demonstrating clinical efficacy in neurodegenerative and genetic CNS diseases.
One of the most advanced classes of RNA therapeutics are antisense oligonucleotides (ASOs), which are short, synthetic single-stranded DNA or RNA molecules that hybridize with complementary RNA targets via Watson-Crick base pairing. Depending on their design, ASOs can recruit RNase H to degrade the RNA strand of a DNA-RNA duplex, modulate pre-mRNA splicing by sterically blocking spliceosome components, or inhibit translation by binding to the 5’ untranslated region (UTR) or initiation codon. Chemically, ASOs are often modified with phosphorothioate linkages and 2’-O-methyl or 2’-O-methoxyethyl sugar modifications to increase nuclease resistance, enhance target affinity, and reduce off-target effects.
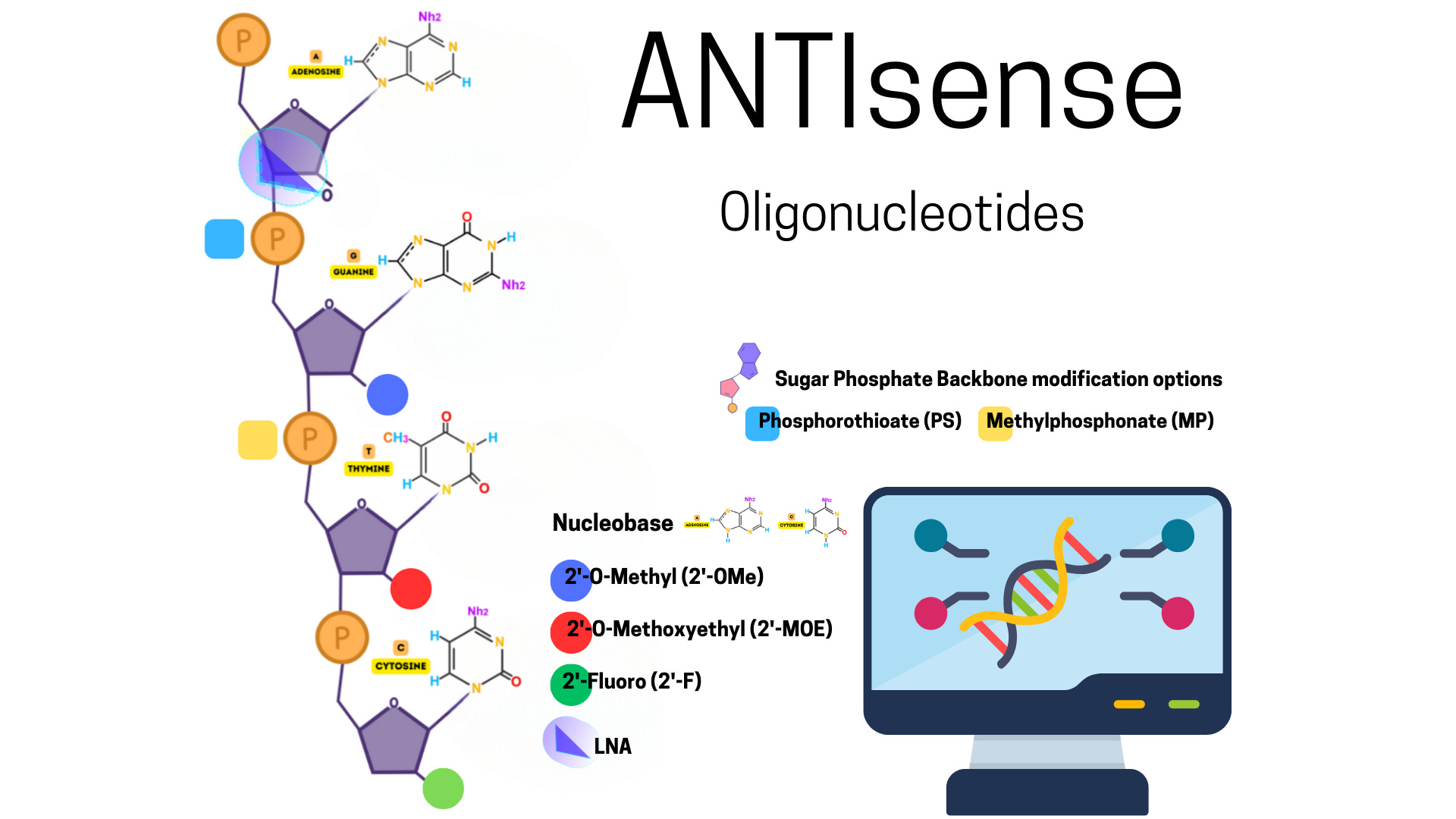
Antisense Oligonucleotides for CNS Therapeutics
Introduction
Antisense oligonucleotides (ASOs) represent one of the most clinically advanced classes of RNA therapeutics. They are short, synthetic, single-stranded DNA or RNA molecules that base-pair with complementary RNA sequences to modulate gene expression. In the context of central nervous system (CNS) disorders, ASOs offer a unique ability to target previously undruggable genes through sequence-specific mechanisms, bypassing the limitations of conventional small molecules and biologics. Their mechanisms of action, chemical properties, pharmacokinetics, and delivery challenges must be precisely engineered for efficacy and safety within the CNS environment.
Mechanisms of Action
RNase H1-Mediated Cleavage
ASOs designed to recruit RNase H1 act through a degradation-based mechanism. These ASOs are typically structured as gapmers, which consist of a central DNA segment that forms the RNase H1 recognition site, flanked by chemically modified ribonucleotides that increase nuclease resistance. Upon hybridization with the target RNA, the DNA-RNA heteroduplex is recognized by endogenous RNase H1, which cleaves the RNA strand while leaving the DNA intact. The ASO remains available for additional rounds of hybridization and cleavage, allowing catalytic turnover of the RNA substrate.
Steric Blockade of RNA Processing
ASOs that do not induce RNA degradation can instead function by physically blocking access to RNA-binding proteins or regulatory complexes. Splice-switching ASOs are designed to bind pre-mRNA at intronic or exonic motifs such as exonic splicing enhancers or silencers. This binding prevents spliceosomal components from interacting with the RNA, thereby modulating exon inclusion or exclusion. Nusinersen, for instance, promotes exon 7 inclusion in the SMN2 gene to restore SMN protein expression in spinal muscular atrophy. Translation-blocking ASOs bind to the 5’ untranslated region or the AUG start codon of mRNAs, preventing ribosomal scanning or assembly and effectively inhibiting protein synthesis.
Design Considerations
Target Accessibility
ASO efficacy depends on the accessibility of the RNA target site. Computational tools such as mFold and RNAstructure are used to predict RNA secondary structures and identify regions likely to be single-stranded and exposed. These predictions are validated through empirical techniques including SHAPE-Seq and DMS-MaPseq, which chemically probe RNA flexibility in vitro or in vivo. Accessible regions are selected to ensure efficient hybridization and minimal interference from RNA tertiary structures or RNA-binding proteins.
Thermodynamic Optimization
Hybridization stability is a critical parameter in ASO design. Melting temperature (Tm) is optimized to fall within the range of 55 to 65 degrees Celsius to ensure that the ASO-RNA duplex remains stable under physiological conditions. Tm is influenced by GC content, nucleotide sequence, oligonucleotide length, and chemical modifications. Binding affinity must be high enough to ensure selective engagement with the intended RNA without significant binding to non-target transcripts.
Chemical Modifications
Backbone Chemistry
Phosphorothioate (PS) linkages are commonly employed in ASO backbones. In this modification, a non-bridging oxygen in the phosphate group is replaced with sulfur, which enhances resistance to exonucleases and endonucleases. PS linkages also promote plasma protein binding, which increases the ASO’s half-life in circulation and facilitates tissue uptake. However, this modification can reduce duplex stability and increase nonspecific protein interactions, necessitating careful optimization.
Sugar Modifications
The 2’-O-methyl and 2’-O-methoxyethyl modifications on the ribose sugar improve ASO stability by enhancing resistance to nucleases and increasing Tm. These modifications also reduce activation of innate immune receptors such as Toll-like receptors 7 and 8. They are typically applied to the flanking regions of gapmer ASOs, where they protect the molecule without interfering with RNase H1 activity. Locked nucleic acids (LNAs), which constrain the ribose ring via a methylene bridge, confer even greater binding affinity and are often used in mixmer configurations to stabilize the ASO-RNA duplex further. Base modifications, including 5-methylcytosine and tricyclo-DNA, are also used in specific contexts to modulate ASO pharmacokinetics and enhance CNS penetration.
Pharmacokinetics and Cellular Uptake
Endocytic Uptake Pathways
ASOs enter cells through multiple forms of endocytosis, including adsorptive uptake, scavenger receptor-mediated internalization, and clathrin- or caveolin-mediated endocytosis. Scavenger receptors such as SR-A1 and SR-B1 are known to mediate efficient uptake of phosphorothioate-modified ASOs. After internalization, ASOs are typically sequestered in endosomes, and only a small fraction escapes into the cytosol or nucleus, which limits bioavailability at the target site.
Tissue Distribution and Half-Life
Phosphorothioate ASOs bind to plasma proteins such as albumin, reducing renal clearance and improving distribution into tissues, including the CNS. Intrathecal administration is the primary route for CNS delivery, allowing direct infusion into cerebrospinal fluid and bypassing the blood-brain barrier. ASOs demonstrate prolonged tissue half-lives in the CNS, ranging from weeks to months, which enables infrequent dosing and long-term gene modulation.
Immunogenicity and Off-Target Effects
Unmodified or poorly designed ASOs can trigger immune responses through recognition by pattern recognition receptors. Toll-like receptors 3, 7, and 8 detect RNA sequences in endosomes, while cytoplasmic receptors such as RIG-I and MDA5 respond to double-stranded or structured RNA. These activations lead to the induction of interferon-stimulated genes and inflammatory cytokines. Chemical modifications are implemented to suppress immune activation and increase tolerability. Off-target effects may arise from partial sequence complementarity to unintended transcripts or nonspecific interactions with RNA-binding proteins, such as those found in nuclear paraspeckles. Such risks are minimized through in silico transcriptome scanning and biochemical screening.
CNS-Specific Optimization
For therapeutic use in neurological disorders, ASOs must be chemically stable in cerebrospinal fluid, exhibit minimal immunogenicity in glial and neuronal populations, and achieve effective distribution across the CNS. Intrathecal and intracerebroventricular delivery routes provide direct access to the CNS, and in some cases, ASOs are taken up by peripheral axon terminals and undergo retrograde transport to reach central neurons. This mechanism is particularly relevant in motor neuron disorders such as spinal muscular atrophy. Designing ASOs for CNS applications involves an integrated strategy encompassing RNA biology, nucleic acid chemistry, pharmacology, and neuroanatomy to ensure therapeutic efficacy and safety.
Small interfering RNAs (siRNAs) represent another widely studied RNA modality that harnesses the endogenous RNA interference (RNAi) pathway. These double-stranded RNAs are approximately 21–23 nucleotides in length and are processed intracellularly to load the antisense strand into the RNA-induced silencing complex (RISC). The RISC-anchored siRNA guides the complex to complementary mRNA, where the Argonaute 2 (Ago2) endonuclease mediates cleavage of the target transcript, leading to post-transcriptional gene silencing. siRNAs provide exquisite sequence specificity but are sensitive to nucleolytic degradation, necessitating protective formulation strategies such as lipid nanoparticles (LNPs) or conjugation to cell-penetrating ligands.

Messenger RNA (mRNA) therapeutics offer a contrasting approach: rather than silencing endogenous gene expression, synthetic mRNAs are introduced to express a therapeutic protein in situ. These mRNA molecules are engineered with modified cap structures (such as anti-reverse cap analogs), optimized 5’ and 3’ UTRs, and poly(A) tails to enhance translation and cytoplasmic stability. Nucleoside modifications—such as incorporation of pseudouridine and N1-methyl-pseudouridine—are frequently used to reduce activation of innate immune sensors like Toll-like receptors (TLRs) and RIG-I. In the CNS, mRNA therapies are being explored for enzyme replacement, neurotrophic factor delivery, and even as vaccines for neuro-oncological applications.
Messenger RNA Therapeutics for CNS Applications
Introduction
Messenger RNA (mRNA) therapeutics represent a distinct class of RNA-based medicines that function not by suppressing endogenous gene expression but by introducing exogenous, synthetic mRNA into cells to direct the in situ production of therapeutic proteins. In the CNS, this approach is particularly attractive for addressing diseases where the underlying pathology stems from a deficit of a functional protein, a need for neuroprotective factors, or the requirement for antigen-specific immunotherapy. The ability to transiently express proteins without genomic integration offers a unique safety and versatility profile. However, the development of mRNA therapeutics suitable for CNS use involves overcoming significant biochemical, immunological, and delivery-related challenges.
mRNA Structure and Engineering
Synthetic mRNAs used for therapeutic purposes are meticulously engineered to optimize stability, translation efficiency, and immunocompatibility. The basic structure of a functional mRNA includes a 5’ cap, a 5’ untranslated region (UTR), a coding sequence (CDS), a 3’ UTR, and a polyadenylated tail. The 5’ cap is essential for efficient ribosomal recognition and protection against exonuclease degradation. Modified cap structures, such as anti-reverse cap analogs (ARCAs), are incorporated during in vitro transcription to ensure proper orientation and increased binding to the eukaryotic translation initiation factor eIF4E. In some designs, CleanCap or other proprietary capping strategies are used to enhance translational yield and reduce recognition by innate immune sensors.
The untranslated regions flanking the coding sequence are also critical. The 5’ UTR is optimized to reduce secondary structure, which facilitates efficient scanning by the 43S pre-initiation complex and improves ribosomal loading. The 3’ UTR is tailored to include stability elements such as AU-rich elements and microRNA binding sites, which influence transcript turnover and localization. The coding sequence itself is codon-optimized to match the host species’ tRNA abundance profile, minimizing translational stalling. Finally, the poly(A) tail, typically consisting of 100 to 120 adenosine residues, protects the transcript from 3’ exonucleases and promotes mRNA circularization via interaction with poly(A)-binding proteins, thereby enhancing translational efficiency.
Nucleoside Modifications and Immune Evasion
Unmodified synthetic mRNA is inherently immunogenic. Exogenous RNA is sensed by innate immune receptors such as Toll-like receptors TLR3, TLR7, and TLR8 in endosomes, and cytoplasmic RNA sensors including RIG-I and MDA5. Activation of these receptors leads to the production of type I interferons and proinflammatory cytokines, which inhibit translation and promote transcript degradation. To mitigate this, mRNA therapeutics employ chemically modified nucleosides that mimic natural post-transcriptional RNA modifications. The most commonly used modifications are pseudouridine and N1-methyl-pseudouridine. These modifications reduce base-pairing rigidity and alter the RNA secondary structure in a manner that reduces receptor binding affinity without compromising ribosomal recognition.
Modified nucleosides also increase mRNA stability by reducing activation of RNases and limiting the recruitment of RNA degradation complexes. During in vitro transcription, modified triphosphates are incorporated directly by RNA polymerase. High-purity mRNA synthesis also involves the removal of double-stranded RNA contaminants that can arise during template mispairing. This is achieved using chromatographic purification techniques such as reverse-phase high-performance liquid chromatography (RP-HPLC), which further reduces immunogenicity and improves translational performance.
Translation and Protein Expression in CNS Cells
Once delivered to the cytoplasm, synthetic mRNA bypasses the nuclear compartment and engages directly with the host translational machinery. The initiation of translation begins with the assembly of the 43S pre-initiation complex, composed of the 40S ribosomal subunit, initiation factors such as eIF2-GTP-tRNAiMet, and additional scaffolding proteins. The complex binds the 5’ cap structure and scans the mRNA until it identifies an AUG start codon within an optimal Kozak consensus sequence. Translation proceeds through elongation, mediated by eEF1 and eEF2, and terminates at a stop codon, after which the protein is released and folded with the assistance of chaperones.
In CNS-targeted applications, cell type specificity of protein expression is determined by the delivery vector and route of administration, not by the mRNA sequence itself. Astrocytes, neurons, oligodendrocytes, and microglia are all capable of translating exogenous mRNA, although efficiency can vary based on cell-intrinsic uptake mechanisms and cytoplasmic conditions. The transient nature of mRNA translation is an advantage in the CNS, where persistent overexpression of certain proteins could lead to excitotoxicity, inflammation, or maladaptive plasticity. The duration of expression is typically on the order of hours to a few days, depending on mRNA sequence, stability elements, and cellular context.
Applications in CNS Disease
mRNA therapeutics are being investigated across multiple domains of CNS pathology. In enzyme deficiency disorders such as certain lysosomal storage diseases, mRNA encoding the missing enzyme is delivered directly to the CNS to restore metabolic function. For example, mRNA coding for β-glucuronidase or arylsulfatase A has been tested in preclinical models of mucopolysaccharidosis and metachromatic leukodystrophy, respectively. Neurotrophic factors such as brain-derived neurotrophic factor (BDNF), glial cell line-derived neurotrophic factor (GDNF), and insulin-like growth factor 1 (IGF-1) have also been encoded into mRNA constructs for delivery to the CNS to support neuronal survival and synaptic plasticity in models of Parkinson's disease, amyotrophic lateral sclerosis, and spinal cord injury.
mRNA-based immunotherapies are also being developed as vaccines for neuro-oncological indications such as glioblastoma. These vaccines encode tumor-specific neoantigens or shared oncogenic antigens to elicit T cell-mediated cytotoxic responses against malignant cells. Delivery to the CNS for these applications may be local or systemic, depending on the immune accessibility of the tumor microenvironment.
Delivery Considerations
Delivering mRNA to the CNS poses substantial challenges due to its size, negative charge, and susceptibility to nuclease degradation. To overcome these barriers, mRNA is encapsulated in delivery vehicles such as lipid nanoparticles (LNPs), which protect the transcript and facilitate endosomal escape. LNPs are composed of ionizable lipids, phospholipids, cholesterol, and polyethylene glycol-conjugated lipids, which self-assemble into vesicles that can fuse with cellular membranes. Surface modification of LNPs with CNS-targeting ligands, such as transferrin or rabies virus glycoprotein-derived peptides, enhances uptake by brain endothelial cells and neurons. Alternatively, direct intrathecal or intracerebroventricular injection allows bypassing of the blood-brain barrier entirely, ensuring access to the cerebrospinal fluid and CNS parenchyma.
mRNA uptake into cells occurs primarily via endocytosis, and efficient endosomal escape is a limiting factor for cytoplasmic delivery. Ionizable lipids in LNPs become protonated in the acidic endosomal environment, promoting membrane destabilization and release of mRNA into the cytoplasm. Other vehicles under development include exosomes, cell-penetrating peptides, and polymeric nanoparticles, each with distinct physicochemical properties that influence biodistribution, half-life, and cell type specificity.
mRNA therapeutics hold significant promise for the treatment of CNS disorders by enabling direct, transient protein expression within the brain and spinal cord. Advances in mRNA engineering, immunoevasive chemistry, and delivery technologies have transformed mRNA from a fragile, immunogenic molecule into a potent therapeutic agent. Applications in enzyme replacement, neurotrophic support, and neuroimmunotherapy are under active investigation, and ongoing work continues to optimize the pharmacodynamics, distribution, and safety of mRNA delivery systems. The integration of molecular neuroscience, RNA chemistry, and targeted delivery science will be critical to realizing the full therapeutic potential of mRNA in neurology.

Other RNA-based strategies include microRNA (miRNA) mimics and inhibitors, as well as long non-coding RNA (lncRNA) modulators. miRNAs are short (~22 nt) non-coding RNAs that endogenously regulate gene expression by binding to complementary sites in the 3’ UTRs of target mRNAs, leading to translational repression or deadenylation and decay. Therapeutic inhibition of specific miRNAs is achieved using anti-miRs or locked nucleic acid (LNA)-modified oligonucleotides. Conversely, mimics can be used to restore miRNA function in disease contexts where endogenous levels are pathologically suppressed. lncRNAs, which exceed 200 nucleotides, modulate transcription, chromatin architecture, and post-transcriptional regulation. Their therapeutic potential is being explored via ASO-mediated degradation, CRISPR-dCas13 systems, and RNA-binding protein modulation.
Regulatory Non-Coding RNAs as Therapeutic Targets in CNS Disorders
Introduction
In addition to messenger RNA and traditional antisense approaches, regulatory non-coding RNAs such as microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) have emerged as potent modulators of gene expression. These RNA species do not encode proteins but exert control over transcriptional and post-transcriptional processes through sequence-specific and structure-specific interactions with DNA, RNA, and proteins. In the central nervous system, where gene regulation is highly dynamic and spatially restricted, the dysregulation of non-coding RNAs has been implicated in a wide range of pathologies, including neurodevelopmental disorders, neurodegeneration, gliomas, and traumatic injuries. Therapeutic strategies aimed at modulating the activity of miRNAs and lncRNAs are now being developed with increasing molecular precision.
MicroRNA Biology and Therapeutic Modulation
Endogenous Function and Mechanism of Action
MicroRNAs are small, non-coding RNAs approximately 20 to 23 nucleotides in length that function as post-transcriptional repressors of gene expression. They are transcribed as primary miRNAs (pri-miRNAs), which are processed in the nucleus by the microprocessor complex composed of Drosha and DGCR8 into precursor miRNAs (pre-miRNAs). These pre-miRNAs are exported to the cytoplasm by Exportin 5 and further processed by Dicer into mature double-stranded miRNAs. The guide strand is then loaded into the RNA-induced silencing complex (RISC), where it directs the complex to complementary sequences typically located within the 3’ untranslated regions of target mRNAs. Depending on the degree of complementarity, the result is either translational repression or recruitment of deadenylases and decapping enzymes, ultimately leading to mRNA destabilization and degradation.
Therapeutic Inhibition Using Anti-miRs
Dysregulation of specific miRNAs in the CNS can lead to aberrant gene silencing or overexpression. Therapeutic inhibition of overactive miRNAs is achieved using antisense oligonucleotides known as anti-miRs. These are single-stranded oligonucleotides that are complementary to the mature miRNA sequence, effectively sequestering it and preventing RISC loading. To enhance binding affinity and stability, anti-miRs are frequently modified with locked nucleic acids, phosphorothioate linkages, and 2’-O-methyl or 2’-O-methoxyethyl ribose modifications. LNAs are particularly useful due to their high thermal stability and nuclease resistance. Anti-miRs can also be chemically conjugated with targeting ligands or encapsulated in delivery vehicles such as lipid nanoparticles to enable CNS delivery. In vivo, anti-miRs have demonstrated the ability to reverse miRNA-driven phenotypes in models of epilepsy, glioblastoma, and neuroinflammation.
miRNA Mimics for Replacement Therapy
In pathological contexts where endogenous miRNAs are downregulated, synthetic miRNA mimics can be used to restore their regulatory functions. These mimics are typically chemically stabilized double-stranded RNA duplexes that resemble the Dicer-generated miRNA product. The guide strand is designed to be preferentially loaded into RISC, while the passenger strand is degraded. Modifications are applied selectively to avoid immune activation and ensure strand bias. Effective delivery to CNS tissue is a major challenge, and approaches such as intrathecal injection, cell-penetrating peptides, or AAV vector expression of pri-miRNA transcripts have been employed to overcome the blood-brain barrier. Successful mimic-based strategies have been demonstrated for miR-124 in stroke recovery, miR-132 in synaptic plasticity enhancement, and miR-29 in neurodegeneration.
Long Non-Coding RNAs in CNS Function and Therapeutics
Functional Mechanisms
Long non-coding RNAs are transcripts longer than 200 nucleotides that regulate gene expression through a diverse set of molecular mechanisms. Unlike miRNAs, which primarily act at the post-transcriptional level, lncRNAs modulate gene expression at transcriptional, epigenetic, and post-transcriptional levels. LncRNAs can act in cis or trans by forming RNA-DNA hybrids, scaffolding chromatin-modifying complexes, sequestering transcription factors, guiding splicing regulators, or altering RNA stability. These activities are often mediated through specific secondary structures or protein-binding motifs within the lncRNA molecule. In the CNS, lncRNAs such as MALAT1, MEG3, and NEAT1 are known to influence neurogenesis, synaptic architecture, and glial cell activation.
ASO-Mediated Knockdown of lncRNAs
LncRNAs that are pathologically upregulated can be therapeutically silenced using antisense oligonucleotides. The ASOs are designed to bind either to the mature lncRNA transcript or to nascent nuclear RNA, leading to degradation through RNase H1-mediated cleavage or steric blockade of functional domains. Because many lncRNAs reside in the nucleus, nuclear localization of the ASOs is an important consideration in their design. This can be achieved by leveraging phosphorothioate backbones and shorter oligonucleotide lengths that favor nuclear retention. ASO-mediated knockdown of lncRNAs such as BACE1-AS, which stabilizes β-secretase mRNA in Alzheimer’s disease, has shown preclinical efficacy in reducing amyloidogenic processing.
RNA Editing and CRISPR-based Modulation
Emerging methods for precise modulation of lncRNA function include RNA-targeting CRISPR systems such as catalytically inactive Cas13 (dCas13) fused to effector domains. These platforms allow for programmable binding to lncRNA sequences, enabling either steric hindrance, recruitment of RNA-modifying enzymes, or visualization. In addition, engineered ADAR enzymes directed by guide RNAs can introduce adenosine-to-inosine editing in lncRNAs, thereby modulating their secondary structure or disrupting functional motifs without altering the genome. These technologies offer temporal and spatial control of lncRNA activity and are being developed for CNS applications requiring highly specific intervention, such as in glioma progression and microglial activation.
Modulation of RNA-Protein Interactions
An alternative strategy involves disrupting the interaction between lncRNAs and their protein partners. Small molecules or oligonucleotides can be used to interfere with RNA-protein complexes that are required for lncRNA-mediated gene regulation. This approach requires detailed mapping of RNA-protein interfaces, often accomplished through crosslinking immunoprecipitation followed by sequencing (CLIP-seq). In the CNS, this strategy is being investigated for targeting lncRNA interactions with polycomb repressive complexes, histone acetyltransferases, and RNA-binding proteins involved in neuroinflammation.
Therapeutic targeting of regulatory non-coding RNAs such as miRNAs and lncRNAs represents a rapidly evolving frontier in RNA-based medicine for CNS disorders. The ability to fine-tune gene expression at multiple regulatory levels provides unmatched versatility for intervening in complex, cell-specific pathologies. miRNA mimics and inhibitors offer precise modulation of post-transcriptional silencing pathways, while lncRNA-targeted approaches allow for control over transcription, chromatin dynamics, and RNA-protein interactions. As the field advances, continued progress in delivery technologies, off-target prediction, and structure-function mapping of non-coding RNAs will be critical to unlocking their full therapeutic potential in neurology.

A particularly promising innovation is RNA editing, which allows direct recoding of transcripts without altering the genomic DNA. The most studied form is adenosine-to-inosine (A-to-I) editing, mediated by adenosine deaminases acting on RNA (ADARs). Inosine is interpreted as guanosine during translation, enabling codon reprogramming. Engineered ADARs or guide RNAs can be used to site-specifically correct pathogenic single nucleotide variants in transcripts linked to neurodegenerative disorders. For instance, dysregulated editing of the Q/R site in the GluA2 subunit of AMPA receptors has been implicated in excitotoxicity in amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease.
RNA Editing Therapeutics in Neurological Disorders
Introduction
RNA editing represents a powerful therapeutic paradigm that enables precise, post-transcriptional modifications of RNA sequences without introducing permanent changes to the genomic DNA. The most extensively studied and therapeutically leveraged form is adenosine-to-inosine (A-to-I) editing, catalyzed by the adenosine deaminase acting on RNA (ADAR) family of enzymes. Inosine, once incorporated into RNA, is interpreted by the translational machinery and by reverse transcriptases as guanosine, effectively recoding the transcript. This mechanism provides a programmable approach to correct single-nucleotide mutations at the RNA level, offering a transient and potentially safer alternative to genome editing platforms such as CRISPR-Cas9. In the central nervous system (CNS), where transcript diversity and isoform regulation are critical for synaptic function and neuronal survival, RNA editing is emerging as a promising therapeutic tool for a range of neurodegenerative and neurodevelopmental diseases.
Endogenous A-to-I Editing and ADAR Enzymes
The ADAR enzyme family consists of three human homologs: ADAR1, ADAR2, and ADAR3. ADAR1 and ADAR2 are catalytically active, while ADAR3 is thought to act as a dominant-negative regulator in the brain. ADAR enzymes bind to double-stranded RNA (dsRNA) regions via their double-stranded RNA-binding domains (dsRBDs) and deaminate specific adenosine residues to inosine within these structures. Substrate specificity is influenced by the local RNA structure, flanking nucleotide sequence, and presence of mismatches or bulges in the duplex. ADAR2 is the primary enzyme responsible for editing of transcripts in the CNS, including the Q/R site of the GluA2 (GRIA2) subunit of AMPA-type glutamate receptors. Editing at this site converts a glutamine codon (CAG) to an arginine codon (CGG) by modifying the central adenosine to inosine. This single nucleotide change dramatically alters the receptor’s calcium permeability, protecting neurons from excitotoxicity.
Therapeutic RNA Editing Platforms
Therapeutic RNA editing strategies seek to reprogram pathogenic or functionally deficient mRNAs by redirecting ADAR enzymes to specific editing sites using engineered guide RNAs or RNA-binding scaffolds. Two general approaches are under development. The first relies on recruiting endogenous ADAR enzymes by expressing antisense guide RNAs that base-pair with the target transcript to form a suitable double-stranded structure. These guide RNAs are often optimized with structural motifs that enhance ADAR binding and position the target adenosine within the enzyme’s active site. The second approach employs exogenous delivery of engineered ADAR fusion proteins, typically consisting of the catalytic deaminase domain fused to an RNA-targeting domain such as Cas13 or lambda N peptide, which recognizes a guide RNA scaffold. This allows programmable and modular targeting of virtually any RNA sequence.
Precise editing requires careful design of the guide RNA to form a stable duplex with the target site, incorporating features such as internal loops or mismatches that promote selectivity. Off-target editing is a key concern, especially given that inosine formation at non-target adenosines can lead to missense codons, alternative splicing, or altered miRNA targeting. To minimize these risks, guide RNAs are often truncated or chemically modified to reduce non-specific hybridization, and RNA-seq–based analyses are employed during preclinical optimization to map the global editome.
Application to CNS Diseases
One of the most well-characterized therapeutic targets for RNA editing in the CNS is the GluA2 subunit of AMPA receptors. In healthy neurons, the Q/R site in the GluA2 mRNA is nearly 100 percent edited, ensuring that AMPA receptors incorporating GluA2 are impermeable to calcium. In conditions such as amyotrophic lateral sclerosis (ALS), Alzheimer’s disease, and ischemic brain injury, downregulation or mislocalization of ADAR2 results in incomplete editing at this site. The resulting unedited GluA2 subunits form calcium-permeable AMPA channels, increasing intracellular calcium load and promoting excitotoxic neuronal death. Therapeutic restoration of A-to-I editing at the GluA2 Q/R site using targeted ADAR systems has shown protective effects in preclinical models of ALS and ischemia.
Beyond GluA2, other CNS-relevant transcripts are subject to ADAR-mediated editing and may serve as therapeutic targets. These include the 5-HT2C serotonin receptor, whose alternative editing isoforms alter G protein coupling and synaptic responsiveness, and transcripts encoding ion channels, neurotransmitter receptors, and synaptic scaffolding proteins. Site-directed RNA editing has the potential to correct disease-causing point mutations in monogenic disorders such as Rett syndrome, Dravet syndrome, or familial epilepsy by recoding mutant codons at the transcript level.
Delivery Strategies for RNA Editing in the CNS
Efficient delivery of RNA editing components to the CNS remains a major technical challenge. For strategies relying on endogenous ADAR recruitment, guide RNAs can be delivered via lipid nanoparticles, adeno-associated virus (AAV) vectors, or chemically modified synthetic oligonucleotides. AAV vectors are especially well-suited for CNS applications due to their ability to transduce neurons and glial cells with high efficiency. Serotypes such as AAV9 and AAV-PHP.B have demonstrated widespread CNS transduction following intravenous or intrathecal administration. For exogenous ADAR delivery, AAV vectors encoding catalytically active ADAR2 deaminase domains fused to RNA-targeting scaffolds have been used to induce site-specific editing in vivo. In all cases, sustained expression and appropriate stoichiometry between the guide RNA and editing enzyme are critical for maintaining specificity and efficacy.
Tissue-specific promoters, cell-type targeting ligands, and inducible expression systems are under investigation to enhance spatial and temporal control of RNA editing activity. Local delivery methods, including intrathecal and intraparenchymal injections, are also being employed in preclinical models to achieve targeted CNS exposure and minimize systemic distribution.
Off-Target Effects and Safety Considerations
Although RNA editing offers a non-permanent alternative to DNA editing, it still carries risks related to unintended modifications of non-target transcripts. Off-target A-to-I editing can result in amino acid substitutions, alternative splicing, or dysregulation of untranslated regions, potentially leading to cellular dysfunction. Additionally, the formation of long double-stranded RNA structures required for editing may activate innate immune sensors such as PKR, RIG-I, or MDA5, leading to translational inhibition or interferon responses. To address these concerns, high-fidelity ADAR variants and optimized guide RNA architectures are being developed to enhance target specificity. Comprehensive transcriptome-wide analyses using RNA-seq and inosine-specific sequencing (e.g., ICE-seq, REDI-seq) are employed during preclinical development to characterize the full editing landscape.
The reversibility of RNA editing offers an additional layer of safety. Because edited transcripts are naturally turned over through mRNA decay and cellular division, the editing effect is transient and non-heritable. This makes RNA editing particularly appealing for CNS disorders that require precision intervention without lifelong genetic modification.
RNA editing through site-directed A-to-I conversion has emerged as a precise and programmable approach for correcting pathogenic RNA sequences in the central nervous system. By leveraging either endogenous or engineered ADAR enzymes, therapeutic platforms can target specific adenosines in mRNAs to reconstitute normal protein function, restore regulatory homeostasis, or bypass deleterious mutations. Applications in excitotoxicity, monogenic neurological disorders, and synaptic signaling modulation are currently under preclinical and early translational investigation. The continued refinement of delivery systems, editing specificity, and transcriptome-wide safety profiling will be essential for advancing RNA editing technologies into clinical practice for neurological diseases.
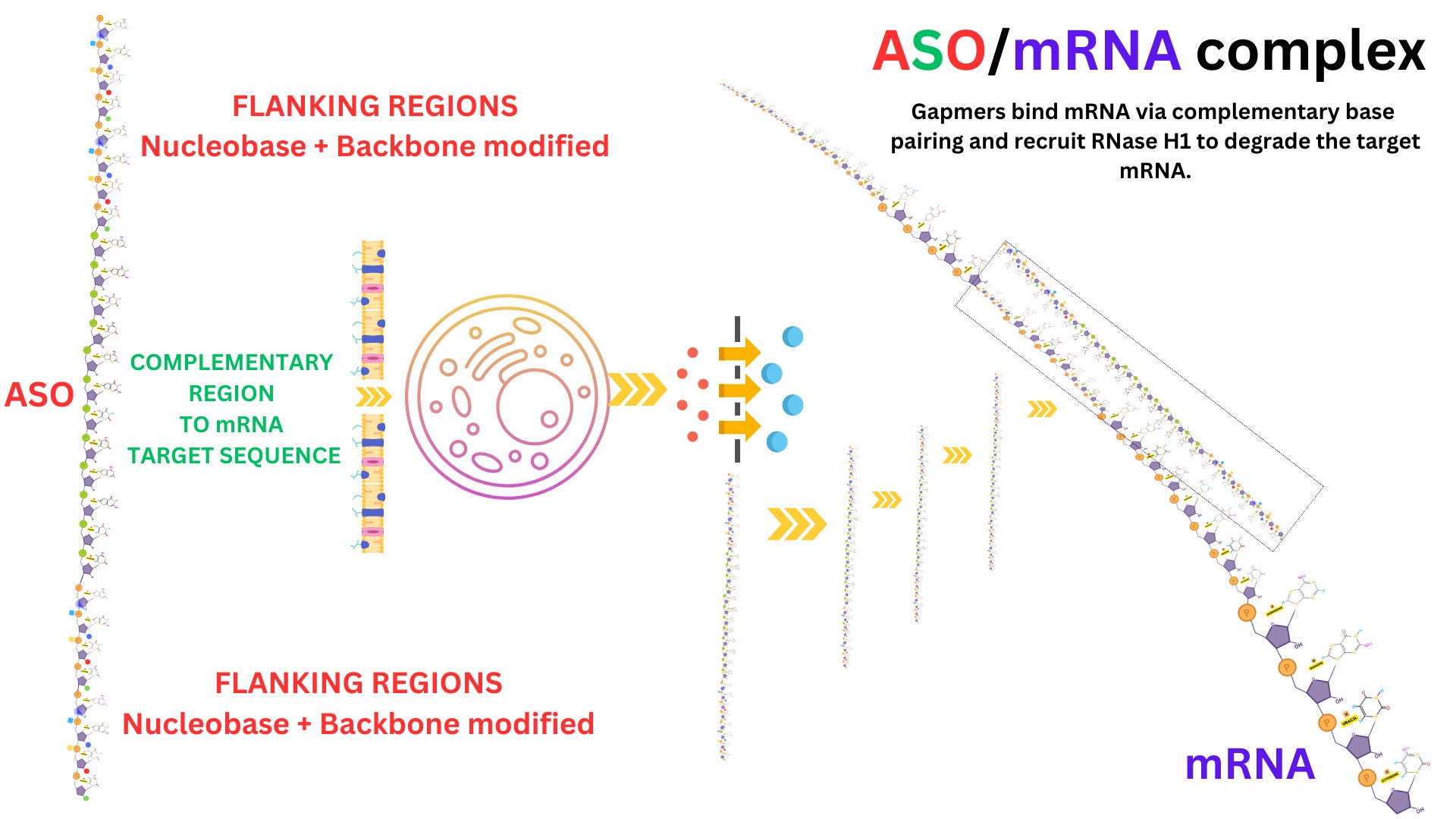
Clinically, RNA-based drugs are already transforming the treatment landscape for CNS diseases. Nusinersen (Spinraza), an ASO targeting the SMN2 gene, was the first FDA-approved RNA therapeutic for spinal muscular atrophy (SMA). It acts by modulating splicing to include exon 7 in SMN2 transcripts, thus increasing the production of functional SMN protein. In Huntington’s disease, ASOs targeting mutant huntingtin mRNA (e.g., tominersen) aim to reduce the levels of the toxic protein aggregate. In ALS, therapeutic efforts are targeting mRNAs encoding TDP-43, SOD1, and FUS using ASOs or siRNAs. Notably, Celosia Therapeutics is developing a viral vector-delivered RNA therapeutic targeting TDP-43 via a CNS-specific promoter to prevent its pathological aggregation.
However, delivery to the CNS remains one of the most significant challenges for RNA therapeutics due to the restrictive nature of the blood-brain barrier (BBB). Several strategies are under active development to address this barrier. Lipid nanoparticles (LNPs) can encapsulate RNA and be engineered with surface ligands that facilitate transcytosis across the BBB. Alternatively, adeno-associated viruses (AAVs), particularly serotypes like AAV9 and AAVrh10, have shown tropism for CNS tissue and are being used for gene and RNA delivery. Intrathecal and intracerebroventricular injections bypass the BBB entirely, allowing direct access to the cerebrospinal fluid and CNS parenchyma. Exosomes and extracellular vesicles are also being engineered as biocompatible carriers for RNA molecules, leveraging their natural ability to cross the BBB and deliver molecular cargo to neurons and glia.
Looking ahead, the development of precision RNA medicines for CNS disorders will hinge on improving the pharmacokinetics, cellular specificity, and long-term safety profiles of these modalities. Advances in computational RNA structure prediction, high-throughput screening for off-target effects, and in vivo imaging of RNA biodistribution are poised to accelerate the next wave of CNS-targeted RNA drugs. As the toolbox for editing, silencing, and expressing RNA in neurons expands, RNA therapeutics are positioned to become a central pillar in the treatment of neurological disease.
The future of CNS Therpeutics
RNA-based therapeutics represent a paradigm shift in the treatment of central nervous system (CNS) disorders, offering an unprecedented degree of specificity, flexibility, and functional diversity compared to traditional pharmacologic and biologic approaches. Unlike small molecules, which often lack target specificity and are limited to modulating surface-accessible proteins, RNA therapeutics can engage virtually any gene product at the transcriptional or post-transcriptional level, including intracellular, nuclear, and previously undruggable targets. Their ability to selectively degrade, splice, edit, or replace RNA transcripts enables precise control over gene expression in both gain-of-function and loss-of-function disease states.
One of the most compelling advantages of RNA therapeutics is their modularity. The functional outcome, whether gene silencing, splicing correction, protein replacement, or sequence correction, can be rapidly tailored by altering the nucleotide sequence or scaffold structure without changing the core chemical platform. This facilitates accelerated development timelines and personalization of therapy, particularly relevant for monogenic or rare neurological diseases where patient-specific interventions are feasible. Moreover, the non-integrative nature of most RNA therapeutics minimizes the risk of permanent off-target genomic alterations, enhancing their safety profile for long-term or reversible modulation of CNS gene activity.
Therapeutic classes such as antisense oligonucleotides (ASOs), small interfering RNAs (siRNAs), messenger RNAs (mRNAs), microRNA modulators, long non-coding RNA agents, and site-directed RNA editing platforms each offer unique molecular mechanisms to address the diverse pathophysiology of neurological disorders. From neurodevelopmental syndromes and motor neuron diseases to neurodegeneration and brain cancers, RNA-based agents are enabling interventions at multiple regulatory nodes, including transcript abundance, splicing fidelity, protein function, and post-transcriptional editing. This multi-tiered therapeutic potential aligns with the complex, systems-level dysfunctions characteristic of many CNS diseases.
Equally transformative are the advances in CNS-targeted delivery technologies. Lipid nanoparticles, engineered viral vectors, chemically stabilized oligonucleotides, and cell-penetrating ligands are overcoming the formidable barriers posed by the blood-brain barrier and neural compartmentalization. Innovations in intrathecal, intraventricular, and minimally invasive systemic delivery are rapidly expanding the anatomical reach and clinical applicability of RNA medicines.
As the field matures, RNA therapeutics are poised not only to complement but to eventually replace conventional CNS drug paradigms. Their precision, programmability, and adaptability uniquely position them to become the standard of care across a broad spectrum of neurological diseases. With continued improvements in delivery, durability, and safety, RNA-based interventions are expected to anchor a new era of neuromolecular medicine, one defined by genetic logic, molecular reversibility, and the potential for curative intervention at the level of gene expression itself.
Super interesting! Thanks.